炎症因素在糖尿病性黄斑水肿中的作用及展望
2022-08-09秦时月徐国旭张敬法
秦时月,徐国旭,张敬法
0引言
糖尿病视网膜病变(diabetic retinopathy, DR)是工作人群视力受损或下降的主要原因,其中糖尿病性黄斑水肿(diabetic macular edema,DME)和增殖性糖尿病视网膜病变(proliferative DR, PDR)是导致患者视力损害的主要原因,患病率分别为23.0%和14.0%[1]。DME是DR最常见的并发症,研究显示若不进行干预治疗,约25%~30%的DR患者3a内将发生具有临床意义的黄斑水肿,导致患者视功能受到严重损害。目前,DME具体发病机制尚未完全阐明,血-视网膜屏障(blood-retinal barrier,BRB)功能受损或破坏所致的渗漏增加是导致DME的主要原因。近年研究表明,炎症因素也参与了疾病的发生与发展[2]。本文旨在阐述炎症因素在DME中作用及机制,以期探究抗血管内皮生长因子(vascular endothelial growth factor,VEGF)和抗炎治疗在DME中的潜在价值。
1炎症细胞的活化
在DME发病机制中,慢性高血糖激活多条生化途径,如蛋白激酶C(PKC)通路、晚期糖基化终末产物(AGEs)增多、多元醇通路、氨基己糖通路、肾素血管紧张系统、聚腺苷二磷酸核糖聚合酶活化等[3-4],导致视网膜缺氧、氧化应激和慢性炎症。小胶质细胞、单核/巨噬细胞、Müller细胞和视网膜色素上皮(retinal pigment epithelial,RPE)细胞等炎症相关细胞活化,释放大量的炎症因子及炎症介质,包括补体系统、VEGF、胎盘生长因子(placental growth factor,PlGF)、肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)、IL-1β、IL-6及IL-8等[5],导致血-视网膜屏障破坏。此外,视网膜血管内皮细胞通过上调细胞间黏附分子-1(intercellular adhesion molecule 1,ICAM-1)及血管细胞黏附分子-1(vascular cell adhesion molecule-1,VCAM-1)表达,导致白细胞黏附和瘀滞(leukostasis),进一步加重视网膜缺氧以及血-视网膜屏障的破坏。
1.1小胶质细胞及单核-巨噬细胞活化小胶质细胞是视网膜中主要的常驻免疫细胞,参与维持视网膜内环境的稳定。一旦视网膜内环境稳态改变,小胶质细胞和单核-巨噬细胞会活化。慢性高血糖和炎症可招募和诱导巨噬细胞和小胶质细胞活化[6-8]。活化的小胶质细胞和单核-巨噬细胞迁移到病变区域,吞噬受损或死亡细胞,导致RPE功能改变,并释放炎症因子[9-10],如VEGF、PlGF、TNF-α、IL-1β、IL-6及IL-8等,并减少视网膜血管附近抗炎介质的产生,加重血-视网膜屏障的破坏[11-13]。在糖尿病大鼠视网膜中,趋化因子配体2/单核细胞趋化蛋白-1[chemokine(C-C motif)ligand 2/monocyte chemotactic protein-1,CCL2/MCP-1]水平升高,招募单核-巨噬细胞浸润[14]。CCL2主要是由小胶质细胞分泌;此外,来源于小胶质细胞的IL-1β也可以诱导视网膜Müller细胞和RPE细胞分泌CCL2[15]。
近年来,光学相干断层扫描(optical coherence tomography,OCT)检测到的视网膜内高反射点(hyper reflective foci,HRF)被认为是预测眼底疾病进展、疗效及预后的新型生物标志物。关于HRF的起源还存在一些争议,有研究认为HRF来源于视网膜内外渗蛋白和/或脂质沉积,被认为是硬性渗出的前体[16]。还有研究认为HRF是血-视网膜屏障破坏的结果,即含脂巨噬细胞迁移到视网膜内[17]。随着血管OCT(OCTA)的广泛应用,近年来HRF被认为可能来源于视网膜内活化的炎症细胞,特别是小胶质细胞和巨噬细胞。活化的小胶质细胞胞体变大,细胞形态呈“阿米巴样”,在OCT中呈现高反射点表现。HRF可见于多种视网膜疾病,包括DR、DME、视网膜静脉阻塞、无脉络膜症、以及其他视网膜退行性疾病[18-20]。在DME患者的OCTA检查中,我们发现视网膜内HRF数量明显增多,主要聚集在视网膜囊样水肿以及网膜下积液的周围,甚至出现在视网膜外层,提示炎症细胞参与了DME的发生与发展(图1)[21]。

图1 DME患者OCTA扫描检测视网膜内HRF(黄色箭头) A:病例1,男,70岁,视网膜内层间积液及视网膜下积液周围出现HRF;B:病例2,女,61岁,视网膜全层及囊样水肿周围出现HRF。
1.2白细胞瘀滞在DME发病机制中,慢性高血糖、氧化应激和升高的VEGF等因素通过上调血管内皮细胞表达ICAM-1诱导单核细胞和白细胞附着到血管壁上并导致白细胞瘀滞(leukostasis)[22-24]。白细胞黏附和瘀滞通常伴随着血-视网膜屏障功能障碍[25-26]。黏附的白细胞还可产生氧自由基、蛋白水解酶及各种炎性因子,进一步加重视网膜血管内皮细胞损伤和血-视网膜屏障破坏[27-28],加重黄斑水肿。采用ICAM-1和CD18中和抗体可抑制白细胞黏附和视网膜血管内皮细胞死亡[29],表明白细胞黏附是导致视网膜血管内皮细胞的死亡和血-视网膜屏障功能障碍的重要原因。
1.3视网膜Müller细胞活化Müller细胞作为视网膜内的大胶质细胞,主要通过内向整流钾离子通道(inwardly-rectifying potassium channel, Kir),特别是Kir4.1和水通道蛋白4(Aquaporin 4, AQP4)[30],将视网膜内的液体和离子排出至视网膜血管或玻璃体腔内,使视网膜处于“相对干”的状态,从而维持视网膜水和离子稳态。任何视网膜应激都可激活RMG,并改变RMG细胞中Kir和AQP4的表达和分布,导致Müller细胞引流功能障碍。此外,Müller细胞也是VEGF的主要来源[31]。活化的Müller细胞分泌VEGF、IL-6和MCP-1[32-33]等炎症因子和趋化因子[34],加重视网膜炎症反应以及血-视网膜屏障破坏。此外,Kir和水通道的异常以及细胞内离子的蓄积还会引起Müller细胞的渗透性水肿(亦称为“细胞毒性水肿或细胞性水肿”),导致Müller细胞排水功能障碍并形成囊样水肿,从而加重黄斑水肿[35]。
临床上,通过OCTA检查,部分DME患者会出现囊样水肿(图2),提示Müller细胞性水肿。在DR中,Müller细胞的Kir4.1分布发生改变,导致视网膜深层血管周围的Kir4.1通道丢失。由于视网膜深层血管中星形胶质细胞缺失,Müller细胞是与视网膜深层血管相互作用的唯一大胶质细胞36,这导致液体积聚在视网膜内核层37,形成囊样水肿。因此,视网膜内囊样水肿通常反映在慢性高血糖和炎症的持续刺激下Müller细胞引流功能障碍,并随病变进展导致Müller细胞性水肿或囊样变性。
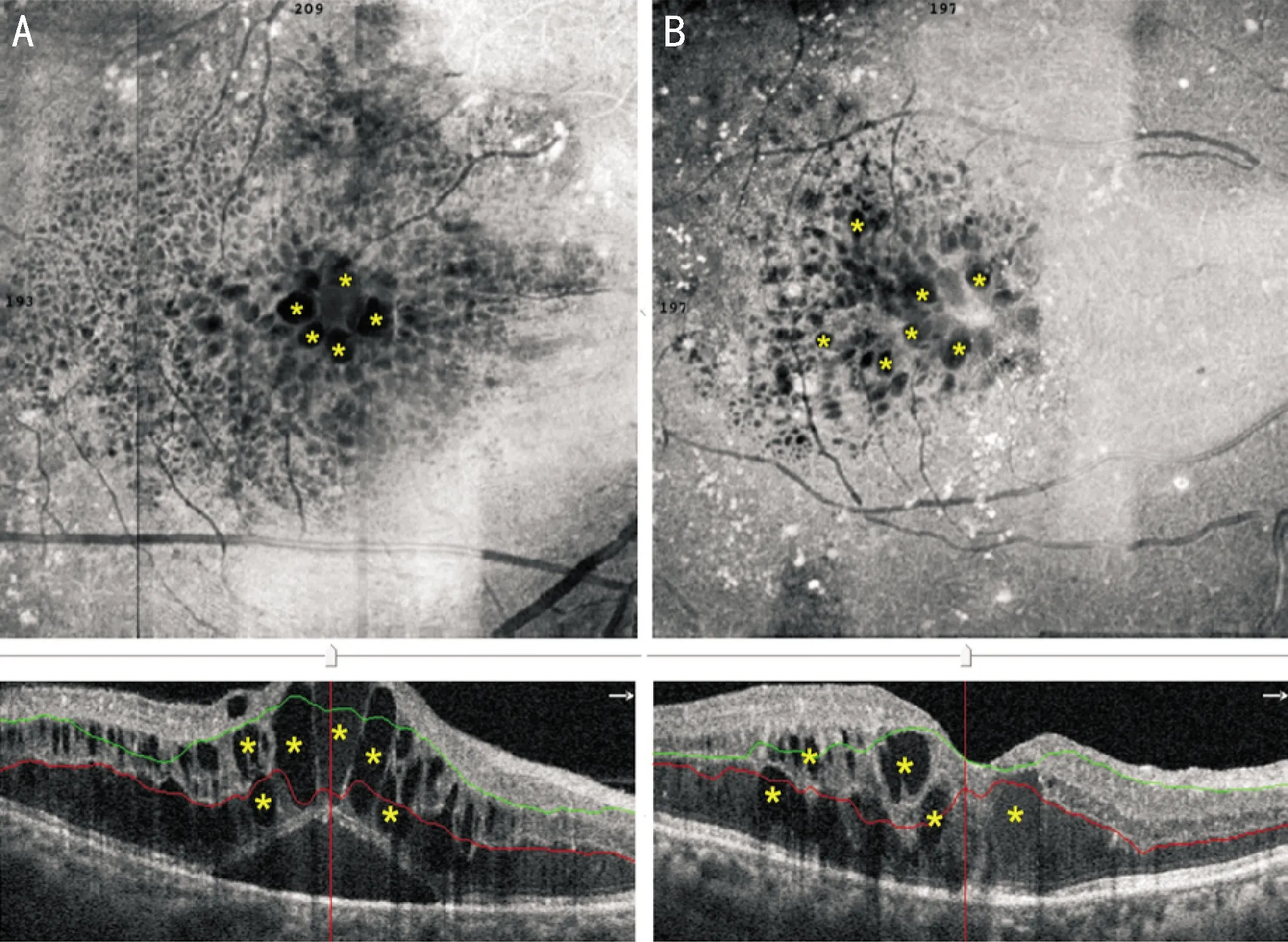
图2 DME患者OCTA检查en-face及B扫描均显示视网膜深层毛细血管层存在大量囊样水肿(黄色星号) A:病例3,男,50岁;B:病例4,女,77岁。
1.4RPE细胞功能障碍RPE除了作为血-视网膜外屏障的重要组成外,还在外层视网膜,尤其是视网膜下液的稳态维持中发挥着重要作用。RPE可被炎症激活,并产生大量的炎性细胞因子和趋化因子,与脉络膜巨噬细胞/小胶质细胞和肥大细胞密切相互作用。视网膜下小胶质细胞的聚集和激活可促进RPE细胞产生促炎介质,进一步加重炎症级联反应[38]和血-视网膜外屏障的破坏[39]。
在部分DME患者中,通过OCT检测可以发现由血-视网膜外屏障破坏或RPE功能障碍导致的视网膜下积液(subretinal fluid,SRF)积聚或中心凹下神经视网膜脱离(subfoveal neuroretinal detachment,SND)(图3)。横断面、前瞻性、病例对照研究表明DME合并SND中HRF数量显著高于无SND的患者[40];此外,回顾性研究发现DME合并SND的患者玻璃体内炎症因子,如IL-6、IL-8表达较非SND患者显著升高[41],提示炎症细胞和炎症因子参与了SND型DME的发病。
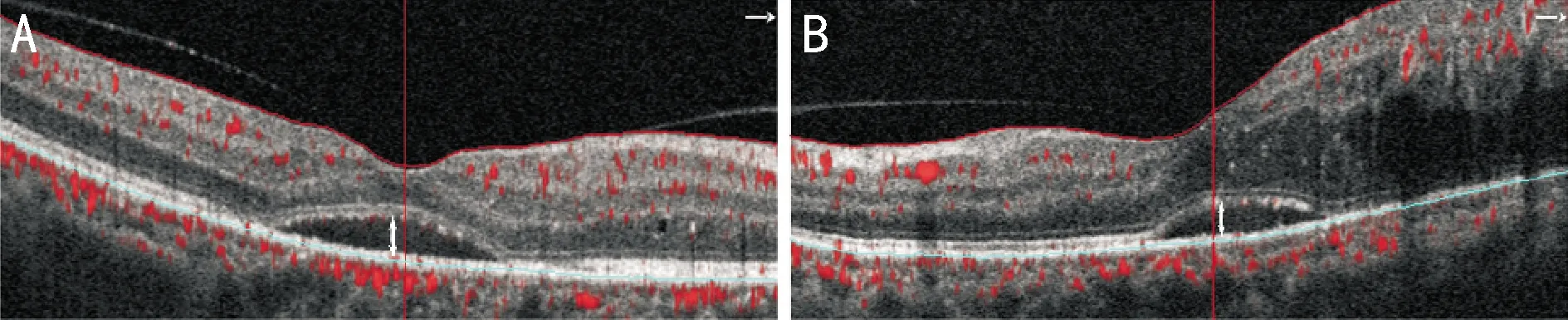
图3 DME患者OCTA检查B扫描均显示黄斑中心凹下SRF(白色双箭头) A:病例5,男,50岁;B:病例6,男,62岁。
作为高度极化的上皮细胞,任何视网膜应激,如糖尿病、炎症,不仅会改变RPE细胞的紧密连接,导致血-视网膜外屏障的破坏;还会导致RPE细胞膜转运体的异常分布,导致RPE细胞引流功能障碍,共同导致SRF形成。DR时,RPE细胞间紧密连接减少[42],水通道蛋白表达模式改变[43]。水通道蛋白对RPE细胞的排水功能至关重要。在缺氧、氧化应激、VEGF和高糖条件下,AQP9在体外培养的RPE细胞中高表达[44]。在链脲佐菌素诱导的1型糖尿病模型中,AQP5、AQP9、AQP11和AQP12在RPE中高表达,而AQP0降低[43]。
综上所述,DME时,多种因素诱导视网膜RMG和RPE细胞活化和功能改变,导致RMG和RPE细胞中水和离子通道的表达和分布异常及排水功能障碍,加重DME。
2炎性因子和炎性介质大量生成和释放
在DME患者的房水或玻璃体中,检测到大量炎性因子和炎性介质,如补体、IL-1β、IL-6、IL-8、VEGF、PlGF、TNF-α、MCP-1、ICAM-1、TGF-β等表达增加,提示炎性因子和炎性介质可诱发炎症反应,加重血-视网膜屏障破坏,参与了DME的发生与发展[45-46]。
2.1VEGF家族以及VEGF受体在DME中的作用VEGF家族成员包括VEGF-A、B、C、D和PlGF,其中VEGF-A与眼部血管性疾病密切相关,可由多种视网膜细胞,如RMG细胞、内皮细胞、RPE细胞、周细胞和星形胶质细胞等产生,这些细胞在高血糖、缺氧[47]、年龄和炎性细胞因子如IL-1β、IL-6等[46]刺激下,上调视网膜内VEGF-A及其他家族成员的表达。VEGF-A通过与VEGF受体(VEGFR-1和VEGFR-2)结合,发挥细胞效应,在血管渗漏、新生血管生成和炎症反应中发挥关键作用[48]。VEGFR-1主要由单核细胞和巨噬细胞表达,与VEGF-A和PlGF特异性结合,刺激炎症因子的产生和单核-巨噬细胞的趋化,在炎症、缺血和癌症等疾病中起关键作用[45]。VEGFR-2主要由血管内皮细胞表达,不仅可以增加血管通透性,还可以通过核因子κB(NF-κB)来调节炎症因子(如MCP-1和ICAM-1)的表达[45]。这表明,除了增加血管通透性外,VEGF还可以直接促进炎症和视网膜内皮细胞的增殖、白细胞黏附和瘀滞。因此,VEGF-A在DME的发病机制中扮演着重要而复杂的角色。
PlGF作为VEGFR-1的主要配体,其同源二聚体仅与VEGFR-1结合。PlGF可有效地促进血管生成,并诱导内皮细胞的生长和迁移[45]。与VEGF-A相似,PlGF还通过刺激单核细胞和巨噬细胞产生炎症因子和趋化因子来调节炎症过程[45]。由于PlGF与VEGFR-1的亲和力高于VEGF-A,因此过量的PlGF可通过结合VEGFR-1置换出已经与受体结合的VEGF-A,参与眼底病变的发生;而置换出的VEGF-A可通过与VEGF-R2结合发挥病理性作用。此外,单体形式的PlGF和VEGF-A可形成异源二聚体,通过与VEGFR-1/VEGFR-2异源二聚体受体结合发挥病理性作用。有研究发现房水中的PlGF水平与DME的严重程度显著相关[49],综上所述,这些发现表明PlGF参与了DME患者黄斑水肿的发病。
2.2补体系统补体系统在黄斑水肿中发挥重要作用,机制与其激活状态有关。例如,C1抑制剂是激肽诱导渗透性的重要调节因子。过敏毒素C3a和C5a通过促使脉络膜中肥大细胞脱颗粒来诱导血管高通透性。此外,C5a通过PI3K和Src激酶途径导致血管内皮细胞收缩并增加细胞旁路的通透性[50]。
2.3TNF-α TNF-α是一种促炎细胞因子,由小胶质细胞、RMG细胞、巨噬细胞、中性粒细胞和T细胞在各种刺激下产生[46]。TNF-α具有多种生物学效应,包括上调黏附分子、增殖、分化和细胞死亡,被认为与DR病变和眼内炎症等密切相关。大量体内外研究表明,在DR病变中,TNF-α主要通过刺激巨噬细胞、小胶质细胞以及其他细胞产生炎性因子,如IL-6、IL-8和TNF-α,增强炎症反应。另一方面,TNF-α可通过激活肿瘤坏死因子受体-1(TNFR-1)和PKCδ/JNK1/2/c-Jun通路诱导ICAM-1的表达,促进白细胞黏附[51],诱导内皮细胞死亡,还可以增加RPE的通透性[46],导致血-视网膜屏障破坏,加重视网膜渗漏和黄斑水肿。
2.4IL-1β IL-1β是一种主要的促炎细胞因子,通常在应对感染、组织损伤或免疫攻击时由单核/巨噬细胞等免疫细胞产生[52]。以往的报道表明,IL-1β可引起视网膜的多种改变,包括白细胞募集、血管通透性增加以及内皮细胞形态和功能的改变[46],进一步破坏血-视网膜屏障,加重视网膜血管渗漏,加重视网膜水肿。IL-1β通过JNK和p38MAPK通路激活NF-κB,促进炎性相关因子和促炎因子的表达,如IL-6、IL-8、MCP-1、TNF-α等[46],导致视网膜炎性反应。
2.5IL-6 IL-6在宿主抵抗环境压力(如感染和组织损伤)中起着关键作用,在刺激下迅速由单核细胞、巨噬细胞和小胶质细胞产生[53],可刺激成纤维细胞、内皮细胞和巨噬细胞产生凝血因子来维持炎症,随后更多的中性粒细胞和巨噬细胞被募集到视网膜,从而导致组织损伤和慢性炎症的持续[46]。IL-6还可直接破坏内皮细胞和上皮细胞的屏障作用,或者通过诱导其他细胞因子和生长因子的产生来增加血管的通透性[54-55],是VEGF介导血管渗漏的重要介质,在DME和其他炎症性眼病的发病机制中起着重要作用[56]。在DME合并SND患者中,玻璃体中IL-6高表达,多元回归分析表明IL-6与SND型DME具有正相关[41]。
2.6IL-8 IL-8是一种促炎趋化因子,通过激活中性粒细胞和T细胞来促进炎症反应,也是新生血管的重要介体。IL-8在DME的发生中起着重要作用,可诱导中性粒细胞和单核细胞聚集[46],此外,IL-8可下调血管内皮细胞间紧密连接蛋白,包括occludin、claudin-5和ZO-1[50],从而增加血管通透性[45],加重黄斑水肿。有研究表明IL-1β可通过p38 MAPK和ERK1/2信号转导通路刺激RMG细胞表达IL-8,进一步增强IL-1β在DR病变中的致病作用[33],这说明在DR炎症反应过程中,不同细胞因子和趋化因子之间存在显著的重叠和相互作用(crosstalk)。
2.7ICAM-1 ICAM-1是一种黏附分子,在视网膜和脉络膜中表达。视网膜缺氧上调ICAM-1的表达[57],ICAM-1通过与白细胞表面的整合素受体结合促进白细胞黏附和瘀滞。ICAM-1介导的白细胞黏附和瘀滞会加重视网膜缺氧和视网膜血管内皮细胞损伤,从而加重血-视网膜屏障的破坏[50]。
3 DME的治疗
DME的发病机制非常复杂,既包括血-视网膜屏障破坏所致的渗漏增加,又包括RMG和RPE细胞功能障碍所致的引流功能减弱。除此之外,炎症因素在DME中也发挥着重要作用,涉及多条生化途径和多种细胞因子,如VEGF、PlGF、炎性相关因子等。
抗VEGF药物的问世使DME的治疗发生了革命性的变化,被认为是当前DME的一线治疗药物。抗VEGF治疗主要通过拮抗VEGF和/或PlGF从而减少上述因子对黄斑水肿的促进作用。大规模的临床试验如RIDE/RISE、DRCR.net Protocol Ⅰ & T和VIVID/VISTA等均显示DME患者经抗VEGF治疗后,视力得到显著改善且黄斑水肿明显减退。
然而,即使经过规范的抗VEGF治疗,只有33%~45%的患者视力有3行或3行以上的改善,大部分DME患者表现为中度应答(5~9个字母改善)或较差的应答(<5个字母改善或更差)[58]。这表明DME发病机制复杂,多种因素参与了其发生与发展。部分患者对抗VEGF药物反应差甚至抵抗的原因可能是由于抗VEGF药物无法靶向除VEGF以外的其他因素导致的病变,如炎性细胞和炎症因子的参与。对于这类患者,可以考虑眼内注射地塞米松玻璃体内植入剂。眼内注射地塞米松玻璃体内植入剂既可以抑制多种炎症细胞,包括小胶质细胞、巨噬细胞、淋巴细胞及视网膜血管内皮细胞,还可以抑制多种炎性因子,如VEGF、PlGF、IL-1β、IL-6、IL-8及TNF-α,从而发挥强大的抗炎效果。
近年来,随着对DR和DME发病机制认识的不断深入,针对新的靶点的药物实验也正在临床前研究和临床试验阶段进行检测和验证中。
Faricimab是双特异性抗体,通过阻断Ang-2和VEGF-A两条途径来治疗多种视网膜血管性疾病。两项针对DME的随机、双盲、多中心、非劣效的全球Ⅲ期临床试验(YOSEMITE和RHINE)表明DME患者经faricimab眼内注射后获得了显著的视力提升和解剖学获益,最长治疗间隔可达16wk。两项临床试验结果均达到其主要终点指标,显示与当前每2mo给药一次的标准治疗方案相比,每4mo给药一次faricimab也可达到非劣效性标准的视力获益[59]。血管内皮蛋白酪氨酸磷酸酶(VE-PTP)是一种内皮细胞特异性受体酪氨酸磷酸酶,可使Tie2失活并抑制其细胞内信号通路。一项Ⅰ期研究表明,DME患者皮下注射VE-PTP竞争性抑制剂可减少黄斑水肿并改善部分患者的视力[60]。趋化因子受体2和5(CCR2/5)位于单核细胞表面,可诱导炎症细胞到达靶组织。在最近的一项Ⅱ期试验中,口服CCR2/5趋化因子受体双拮抗剂可改善部分DME患者视力,但疗效低于玻璃体腔内注射雷珠单抗[61]。一项小型Ⅲ期研究表明,与安慰剂相比,静脉注射英夫利昔单抗(抗TNF-α单克隆抗体)能显著改善DME患者的视力;但对顽固性DME,拮抗TNF-α可能会引起严重的眼内炎症反应[62]。靶向IL-6及其受体的临床研究也在开展中,旨在评估IL-6及其受体的抑制在眼底血管性疾病中的疗效[63]。血管黏附蛋白(VAP)-1是一种类似于ICAM-1的内皮黏附分子。在一项Ⅱ期临床研究中,比较了口服VAP-1抑制剂ASP8232(Astellas, Northbrook, IL, USA)与玻璃体腔注射雷珠单抗对DME患者的疗效[64]。然而,尽管ASP8232几乎完全抑制了血浆中VAP-1的活性,但对DME未见明显改善,且联合治疗与单用雷珠单抗相比没有优势。针对新靶点的研发有望为DME治疗提供更具针对性的靶向治疗措施。
当病情加重或复杂时,需考虑综合治疗方案,如积极控制血糖,眼内注射抗VEGF药物、缓释地塞米松植入物、激光光凝、微脉冲、甚至玻璃体切割术等,最大程度的促进黄斑水肿的吸收。
4总结
尽管目前有各种有效的治疗方法,但DME仍然是工作人群中最常见的致盲原因,因此是一个重大的社会经济负担。DME的发病机制非常复杂,且多因素参与,如血-视网膜屏障破坏、RMG和RPE细胞引流功能障碍、微血管瘤的渗漏、玻璃体黄斑界面因素、玻璃体炎性储库作用,以及炎性因素等。近年研究表明,炎症因素在DME中发挥关键作用,大量的炎症细胞活化和炎症因子释放,导致血-视网膜屏障功能受损,引起视网膜血管通透性增加,甚至刺激RMG和RPE细胞导致细胞引流功能障碍,形成并加重黄斑水肿(图4)。基于这些机制,目前眼内注射抗VEGF药物和抗炎药物(如缓释地塞米松植入物)是DME的主要治疗手段。但是DME患者需要频繁的玻璃体腔注射,而且仍然有相当数量的患者对抗VEGF药物和激素治疗反应不佳。因此,我们需要对DME的发病机制进行更深入的研究,从而研发更精确的靶向治疗药物。比如对全身和玻璃体的生物标记物(microRNAs、蛋白质组学、代谢组学等)的更多研究可能有助于发现其他适用于药物开发的新靶点。此外,通过对DME患者眼内液,如房水或玻璃体液,进行检测和分析,明确主要的致病因子或组合,从而进行更个性化的精准医疗。因此,提高对DME发病机制的认识和理解,寻找新的治疗策略,将是预防和干预DME的关键。
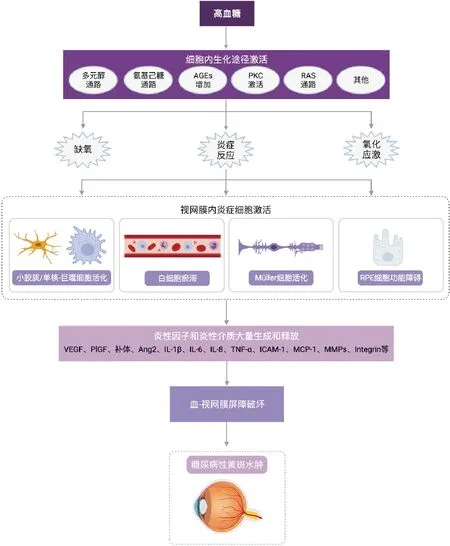
图4 DME发病机制 慢性高血糖激活不同的生化途径,导致视网膜缺氧、氧化应激和慢性炎症反应。炎症细胞激活,炎症因子和炎症介质大量释放,导致血-视网膜屏障破坏,从而导致DME。
