霍奇金淋巴瘤合并免疫性血小板减少症1例并文献复习
2022-08-08杜晨霄张宇卉胡耐博滕广帅邵宗鸿白洁
杜晨霄,张宇卉,胡耐博,滕广帅,邵宗鸿,白洁
天津医科大学第二医院血液科,天津 300000
霍奇金淋巴瘤(HL)是淋巴瘤特殊亚型,其发病过程被认为与免疫因素相关,免疫功能失调可先于疾病,可为独立自发,亦可出现在HL 治疗之后。免疫性血小板减少症(ITP)为一种良性获得性自身免疫性疾病,其发病原因被认为与抗血小板自身抗体、T 细胞异常、血小板相关抗原、病毒感染等相关。两种疾病经规范血液科治疗预后一般良好。但霍奇金淋巴瘤合并免疫性血小板减少症的病例极其罕见,国内尚无报道。国外不同中心研究表明,在HL 患者中合并 ITP 的概率为 0.95‰[1-3],患者常因血小板严重减少、重要脏器出血而失去治疗机会甚至死亡;部分研究者认为ITP 或作为HL 一种罕见的副肿瘤综合征存在,对HL 的发生、预后及随访有提示意义。因此,明确HL 合并ITP 诊断,阐明发病机制,进而寻找正确的治疗尤为重要。
1 病例资料
患者男,70 岁,因皮肤黏膜出血2 周、消化道出血1 周于2020 年9 月就诊于消化科,初诊血常规白细胞8.15×109/L,HGB 59 g/L,PLT 2×109/L,应用皮质激素、免疫球蛋白及输注血小板治疗后疗效欠佳。转入血液科,全身浅表淋巴结B超示:双侧腹股沟淋巴结、双侧腋下淋巴结皮质略增厚;左侧颈部Ⅱ区肿大淋巴结;腹部B 超未见脾肿大。CT 检查示肺部感染病灶。乙肝病毒DNA 定量阴性。幽门螺杆菌抗原测定阴性。于2020 年9 月24 日行胸髂骨髓穿刺,髂骨骨髓活检,骨髓形态学示:双部位均可见巨核细胞明显增多伴产板不良,红系比例增高,碳核样红细胞易见,成熟红细胞体积偏小,中央淡染区扩大。骨髓病理活检报告示:骨髓增生较活跃,粒红巨三系造血细胞增生;未见原始细胞增多;未见异常淋巴细胞明显增多;未见寄生虫、真菌感染及肉芽肿形成;未见转移瘤细胞;未见纤维组织明显增生,见图1。骨髓免疫分型:可见异常B淋巴细胞,FSC及SSC偏小,占有核细胞5.25%,不表达CD103,为CD5-CD10-单克隆B 淋巴细胞。结合以上结果,在除外细菌、病毒感染,实体肿瘤,造血原料缺乏、先天性因素导致血小板减少后,暂考虑诊断为ITP,缺铁性贫血。患者骨髓免疫分型可见异常B 淋巴细胞,其免疫分型不能除外B 细胞淋巴瘤可能,暂诊淋巴瘤待除外。因患者合并消化道出血、高血压、糖尿病,治疗以免疫球蛋白为主,间断予糖皮质激素。患者存在缺铁,予蔗糖铁静脉补铁。患者肺部存在感染,予美罗培南、卡泊芬净抗感染治疗,后复查感染指标及肺部CT,感染好转。经上述治疗,患者血红蛋白上升,但血小板无明显增长。
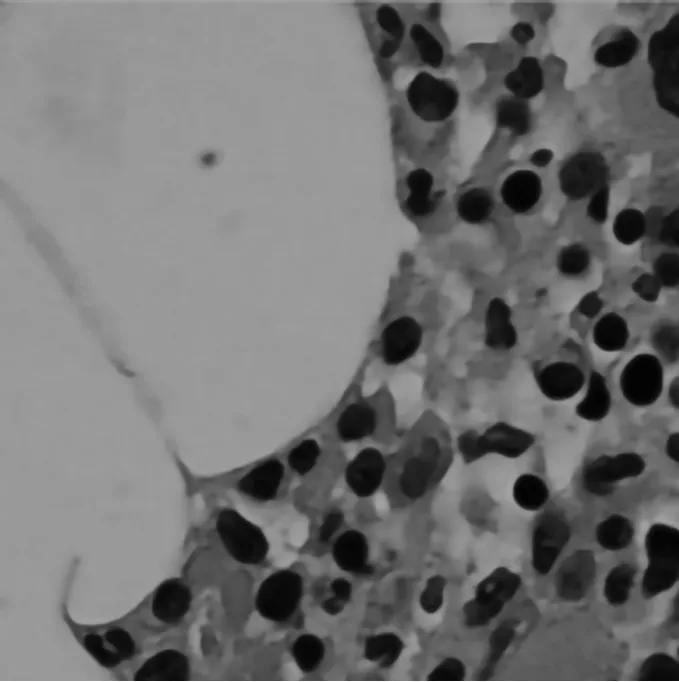
图1 初诊骨髓活检
为进一步寻找病因,于2 周后复查淋巴结B 超,结果示:左侧颈部Ⅳ区及Ⅴ区多发肿大淋巴结,较大者27.9 mm×11.2 mm,双侧腋下、腹股沟淋巴结未见异常,腹主动脉及双侧髂血管周围未见异常及肿大淋巴结。后患者于2020-10-13输注单采血小板后行肿大淋巴结切检术,病理回报示(左颈部淋巴结):结合免疫表型符合经典型霍奇金淋巴瘤,淋巴细胞为主型。免疫组化结果:CD20-,CD3+,CD10-,CD15 残存滤泡+,CD21 残存滤泡+,CD30 散在大细胞+,Ki67(约40%),BCL-2+,BCL-6-,EMA-,PAX-5少量弱+,CXCL13-,CD2+,CD5+,CD7+,CD56 散在+,GranzymeB 散在+,TIA-1散在+,EBER-,大细胞CD30+、MUM1+、Fascin+、CD45-,PAX5弱+,CD20-,CD3-,CD5-,Ki67 大细胞+,CD21FDC+,见图2。行PET-CT检查示:结合病史,左侧颈部活检术后改变,左侧胸锁乳突肌深面、锁骨上区及颈根部多发淋巴结伴代谢增高,结合外院病理结果,考虑淋巴瘤浸润,淋巴结最大者约2.5 cm×1.6 cm,PET 示上述病灶FDG摄取不同程度增高,SUV最大值约4.0。
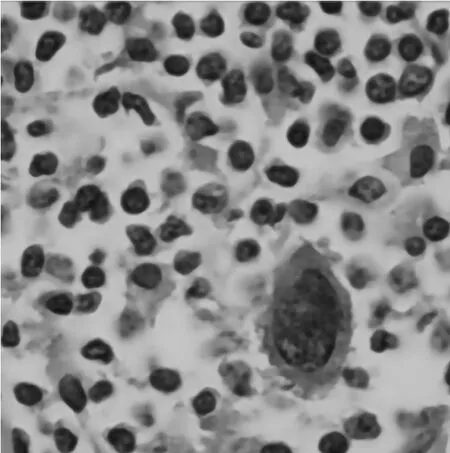
图2 初诊淋巴结组织活检及免疫组化
结合上述诊疗过程及检查结果,患者明确诊断为霍奇金淋巴瘤淋巴细胞丰富型(Lugano 分期Ⅱ期,IPS 评分5 分预后不良),免疫性血小板减少症单克隆 B 细胞待追踪(B 细胞淋巴瘤?)。于 2020 年11 月1 日予多柔比星25 mg/m2、博来霉素10 mg/m2(第1 天和第15 天)、长春新碱2 mg(第1 天和第15天)、达卡巴嗪375 mg/m2(ABVD)方案治疗1个周期后,患者血小板恢复至正常水平,HL 达到CR。患者4周期ABVD方案化疗后行骨穿检查,共检测有核细胞480 000个,其中CD19+成熟B 淋巴细胞占有核细胞0.01%,B 组细胞占有核细胞0.65%,浆细胞占有核细胞0.22%,未见异常B 淋巴细胞(灵敏度0.01%)。随访到发病后12 个月,未见ITP 及HL复发。
2 讨论
HL 合并 ITP 于 1980 年首先被外国学者报道[1],虽然ITP 为非霍奇金淋巴瘤中较为常见的伴随症状,但在霍奇金淋巴瘤中却很少观察到,尤其是自身免疫反应先于HL 发生的病例,国内外罕见。当疾病发生时,HL 往往被ITP 的急性出血症状掩盖,从而失去最佳诊疗时期,诊断困难,同时使已经免疫功能异常患者的临床管理复杂化。ITP 是否为HL 的伴随症状,或为副肿瘤综合征的一种,目前国内外尚未有明确定论。国外多中心研究表明,ITP 合并HL的发生率约占HL 患者的0.95‰,与多数确诊时无自身免疫性细胞减少的患者相比,这些患者发病时中位年龄较大,更常见于疾病晚期,且组织细胞学特征多为混合细胞型,提示HL 恶性程度可能与免疫疾病的发生呈正相关[2]。ABVD 联合化疗(阿霉素、博莱霉素、长春碱、达卡巴嗪)可有效控制HL,减少肿瘤细胞诱导自身免疫状况,从而治疗ITP。
关于HL 合并ITP 的病因及发病机制有多种学说。DIMOU 等[3]回顾合并免疫性血细胞减少的HL患者后指出,混合细胞亚型、疾病晚期、确诊中位年龄较大、多种免疫疾病合并症为HL 合并ITP 的危险因素,且HL 合并ITP 患者的血沉更快、外周血淋巴细胞计数更低[4]。这是由于免疫性血小板减少症的其他潜在疾病包括不同的淋巴增殖性疾病(淋巴瘤、慢性淋巴细胞白血病)、各种自身免疫性和胶原疾病(红斑狼疮、甲状腺疾病、抗磷脂综合征)和部分慢性感染(HIV、丙型肝炎)。在HL合并ITP中,由于自身抗体介导血小板被破坏,加速血小板清除,而抗血小板抗体(PA-IgG)被认为是由于HL 细胞产生,其作用于血小板膜的已知靶点糖蛋白GPIb 和糖蛋白Ⅱb/Ⅲa 上,破坏血小板。因此,可以认为HL 肿瘤活性越高、恶性程度越大,其产生的抗血小板抗体越多,进而导致血小板破坏程度越大。而ITP 与HL 常并发或之后发生,表明两种疾病的发生有免疫功能失衡这一共同发病基础。研究发现,免疫性血细胞减少中的免疫反应是由于对自身抗原的中枢和外周耐受性的破坏导致,前者是通过在骨髓和胸腺的免疫系统成熟过程中消除自身反应性免疫效应物而发生的负选择,包括 B 细胞和 T 细胞耐受[5-6]。在与自身抗原相互作用后,B 细胞通过凋亡和/或能量缺乏诱导发生克隆性缺失。同样,T 细胞在与自身抗原和MHCⅠ类和Ⅱ类抗体相互作用后亦可能产生耐受性。此外,它们可能会转化为能抑制自身反应性T细胞的调节性T细胞(T-reg)。T-reg通过下调白细胞介素(IL)-2和分泌抑制/耐受性细胞因子IL-10和转化生长因子(TGF)-β发挥作用。外周耐受性包括复杂的免疫途径,这些途径可能使残留的自身反应性T 细胞从初级淋巴器官(胸腺和骨髓)排出后失活,并通过克隆缺失、能量诱导或激活Treg 再次发生。自身免疫型血细胞减少的其他关键调节因子是抗原呈递细胞(APCs),有助于Treg 转化及与T 细胞受体相互作用。而在恶性血液病中,特别是淋巴细胞增殖性疾病(LPD)中,自身免疫的上述发病机制可能被放大,其中T 和B 细胞由于高度功能失调,诱导肿瘤发生[7-8]。
本研究报道病例初诊骨髓免疫分型提示存在一组不表达CD103,且CD5-、CD10-单克隆B 淋巴细胞(5.25%),鉴于治疗本病时应用ABVD 方案化疗有效,后随访复查未见其他不同表型淋巴瘤的发生,暂被认为是一组免疫介导单克隆B淋巴细胞。由于HL所致机体免疫功能与致病抗原相互作用后,导致一组B淋巴细胞克隆异常,呈现单克隆表型,进而引发免疫因子风暴,导致其攻击造血细胞,引起血小板减少;同时,此免疫分型结果支持或因B细胞功能长期高度失调控,免疫筛选出恶性克隆细胞,最终导致HL 发生。但是否患者诊断HL 同时存在恶性B 细胞淋巴瘤可能,目前尚未找到证据。而免疫因素与霍奇金淋巴瘤之间因果关系,仍需进一步研究。本例患者免疫调节治疗效果不佳,经过抗肿瘤化疗后HL及ITP 均好转,复查骨髓免疫分型,未再见表型单克B 细胞群,或可提示HL 的发生先于ITP。但由于患者初诊时电泳检测未见异常,骨髓免疫分型提示一群异常单克隆B 淋巴细胞,仍需警惕B 细胞淋巴瘤的发生,在随访过程中需定期复查骨髓免疫分型、血尿固定电泳、蛋白电泳,动态监测全身淋巴结变化。
HL 合并 ITP 的临床表现与 HL 及 ITP 的临床表现相同,HL 临床常见症状有特征淋巴结无痛性肿大,伴或不伴有发热、盗汗、发力、消瘦等B 症状,且已知有诸多罕见相关症状,包括瘙痒、乙醇引起的疼痛、溶血性贫血、溶骨性病变、ITP 等,被认为是副肿瘤综合征[9],其是发生在远离肿瘤或转移病灶地方的恶性肿瘤表现,不是来自肿瘤与周围组织的物理相互作用,而是继发于异位激素作用或自身免疫抗体的产生,或恶性肿瘤产生的其他因素影响。副肿瘤自身免疫综合征可能出现在癌症诊断之前或伴随诊断,也可能在癌症治疗后被视为复发的标志。国外文献回顾显示,有ITP 发生在HL 诊断前后、经HL治疗后很快达到缓解的病例,与副肿瘤综合征的发生及转归过程一致[10-11]。因此,将ITP作为副肿瘤综合征一种,或促使对潜在的恶性肿瘤进行调查、随访[11-12]。而ITP 则可出现皮肤、黏膜,重要脏器的出血,血小板输注效果不佳,常规ITP 治疗效果常较差。临床上HL 合并ITP 患者常以皮肤黏膜、脏器出血为首发症状,增加诊断难度。同时,HL 患者可有多种免疫合并症,LANDGREN 等[12]回顾 7 476例 HL患者指出,类风湿关节炎和系统性红斑狼疮与HL的风险密切相关[13],结节病的个人和家族病史与HL风险增加呈正相关,此类患者亦常合并自身免疫性肾脏病、自身免疫性肝炎、Ⅱ型糖尿病等。HL 合并ITP 的诊断临床上并不困难,主要通过骨髓形态学、组织活检、免疫分型、分子生物学、细胞遗传学结合影像学检查加以确诊,但诊断此类疾病时需通过检查结果与骨髓纤维化、淋巴瘤骨髓侵犯、淋巴瘤化疗相关血小板减少、原发免疫性血小板减少症等鉴别诊断[8]。
HL 合并ITP 的发病罕见,现有文献报道为病例系列或个案分析,缺乏前瞻性研究,且患者淋巴瘤类型、分期、ITP 严重程度、并发症及基础健康情况存在异质性,故尚无推荐治疗方案。DIMOU 等[3]研究合并ITP及AIHA的HL病例指出,ITP严重程度随肿瘤负荷增加而加重,随淋巴瘤病情缓解亦可减轻,因此治疗多以HL 本病治疗为主。经典的ITP 治疗方案,如糖皮质激素和静脉注射免疫球蛋白(IVIG)及血小板抗体靶向治疗,在既往病例中疗效较差,或血小板计数无应答,或血小板回升后又很快下降。而接受ABVD(阿霉素、博莱霉素、长春碱、达卡巴嗪)联合化疗治疗原发性淋巴瘤,均能有效控制淋巴瘤的基础疾病和免疫状况。HL 继发ITP 对化疗的反应迅速、持久,而对IVIG 及激素治疗无效。80%患者自身免疫状况在ABVD 的前半个周期(周期Ⅰa)后好转,且HL 达到CR,ITP 无复发记录。而利妥昔单抗为抗CD20单克隆抗体,通过快速消除体内B细胞,广泛用于治疗各种抗体介导的自身免疫性疾病,包括AIHA、ITP、Evans 综合征等免疫性血细胞减少症[14]。HL 合并 ITP 时,利妥昔单抗能同时起到抗肿瘤及免疫廓清的双重作用,联合化疗可提高HL 合并ITP 患者缓解率,降低复发率,延长无进展生存率及生存时间,目前已成为霍奇金淋巴瘤合并AIHA、ITP 和Evans 综合征等抗体介导免疫性血细胞减少的一线方案治疗。
综上所述,HL 合并ITP 病例罕见,临床患者常以皮肤黏膜、脏器出血为首发症状,HL 的症状被掩盖,诊断难度增加,且患者常合并多种免疫疾病。其发病机制主因免疫机制相互作用导致。HL合并ITP的诊断依赖淋巴结及骨髓活检,需与淋巴瘤骨髓浸润、骨髓纤维化等相鉴别,以ITP 为首发症状的HL治疗上应及时给予抗肿瘤的治本治疗,积极、规律的ABVD 联合化疗对本病效果较好,CD20 单克隆抗体可起到免疫清除及抗肿瘤作用。
