新生儿22q11.2缺失综合征5例临床及基因分析
2022-08-08崔清洋杨勇晖胡劲涛桑桂梅孙亚洲贺晓日
崔清洋,杨勇晖,胡劲涛,桑桂梅,孙亚洲,李 雯,贺晓日
(1. 新乡医学院第一附属医院 儿科,河南 卫辉 453100;2.中南大学湘雅二医院儿童医学中心 新生儿专科,湖南 长沙 410011)
22q11.2缺失综合征(22q11.2 deletion syndrome,22q11.2DS)是由拷贝数变异所造成的常染色体显性遗传的相邻基因缺失综合征,该病是引起先天性心脏病(congenital heart disease,CHD)的第二大常见原因,仅次于唐氏综合征[1],在活产儿发生率为1/4 000~1/6 000[2],可涉及多种表型异常,临床容易漏诊及误诊。20世纪60年代Angelo DiGeorge首次报道了DiGeorge综合征,20世纪70年代Robert博士报道了腭心面综合征,1978年日本的一组心脏病学家将其称之为圆锥干异常面部综合征。因这些综合征的大多数患者均有共同的基因缺陷即22q11.2缺失,故目前统称22q11.2DS[3]。22q11.2DS临床特征为先天性甲状旁腺功能减退和胸腺发育不良所致的细胞免疫缺陷,给家庭及社会带来沉重负担。本研究通过报道新乡医学院第一附属医院新生儿科及中南大学湘雅二医院新生儿专科通过基因确诊的5例22q11.2DS新生儿病例,旨在加强对该病在新生儿期的认识。
1 资料与方法
1.1病例选择 选择2018年2月-2021年10月在新乡医学院第一附属医院新生儿科和中南大学湘雅二医院新生儿专科住院治疗、临床表型怀疑为22q11.2DS的新生儿患儿。怀疑为22q11.2DS的新生儿临床表型有:胸腺小或缺如,CHD,反复低钙血症,感染不易控制。本研究所有检查征得患儿家属同意。
1.2方法
1.2.1资料收集 收集患儿一般资料和临床资料(临床表现、实验室及影像学检查结果、治疗、结局等)。
1.2.2基因检测 血样送至医学检验所进行P250 MLPA试剂盒检测。首先使用Qiagene试剂盒提取样本基因组,之后进行样本质检,质检合格后的样本经DNA变性、杂交、连接、聚合酶链式反应(PCR)等步骤,获得扩增后样本,使用ABI 3130仪器对扩增后样本进行毛细管电泳,使用Coffalyser软件对下机数据进行分析,并与参考序列进行比对获得最终检测结果。
2 结 果
2.1一般资料 共纳入22q11.2DS 5例,产前诊断1例。男性3例,女性2例。早产儿2例,剖宫产4例。由产科出生后立即转入3例,出生后13天及1月17天(矫正胎龄40+5周)由外院转入2例。父母均非近期结婚,无可疑家族史。
2.2临床资料 ①病例1,男,因咳嗽伴喉中痰鸣2天入院。外院及本院心脏彩色超声均为室间隔缺损(VSD)并房间隔缺损(ASD),入院后感染不易控制,先后予美洛西林钠舒巴坦、头孢哌酮舒巴坦及美罗培南抗感染。电解质无异常。细胞免疫:CD3+为1 704×106/L[正常值范围:(2 300~6 450)×106/L],CD4+为1 016×106/L[正常值范围:(1 690~4 699)×106/L],CD8+为670×106/L[正常值范围:(720~2 000)×106/L],均降低。1月13天外院予VSD修补术+ASD修补术+动脉导管结扎术,术中发现胸腺缺如。目前患儿生长发育落后。②病例2,男,因胎儿期发现左肾积水、试管婴儿入院。其母亲曾于某省保健院羊水穿刺提示染色体22q11.21区段检出致病性3.152 Mb缺失片段。住院第2~3天出现发热,先后予头孢他啶、美罗培南、头孢哌酮舒巴坦联合利奈唑胺抗感染,胸部CT提示胸腺发育不良,淋巴细胞亚群CD3+:43.96%(正常值范围:60.00%~84.00%),CD3+CD8+9.08%(20.00%~35.00%),CD3+CD4+32.16%(36.00%~55.00%)。住院第5天出现低钙血症,钙离子波动于1.19~1.72 mmol/L,25(OH)D3为9.1 ng/ml,降钙素及甲状旁腺素(parathyroid hormone,PTH)均无异常,补充钙剂及维生素AD滴剂后上升缓慢,肌注维生素D2后逐渐上升至正常。目前患儿生长发育落后。③病例3,女,因胎龄35+3周、出生后面色发绀13 min入院。母孕期四维彩色超声提示胎儿为法洛四联症。胸片提示胸腺小。电解质无异常。淋巴细胞亚群示:CD3+:53.65%(正常值范围:60.00%~84.00%),CD3+CD8+15.90%(正常值范围:20.00%~35.00%),CD3+CD4+34.70%(正常值范围:36.00%~55.00%);出生5月后外院予手术治疗,目前患儿生长发育落后。④病例4,女,因发现胎儿期心脏畸形4月余,生后口吐泡沫、发绀35 min入院。入院时钙离子1.88 mmol/L,胸腺彩色超声发现胸腺缺如。住院1周后患儿出现低钙血症,钙离子波动于1.47~1.28 mmol/L,予钙剂及维生素D对症治疗后效果欠佳。IgG: 6.35 g/L(正常值范围:8.60~17.40 g/L),IgA:0.01 g/L(正常值范围:1.00~4.20 g/L),IgM:0.07 g/L(正常值范围:0.50~2.80 g/L),PTH 0 min:4.30 pg/ml(正常值范围:18.50~88.00 g/L),淋巴细胞亚群示:CD3+:65.00%(正常值范围:56.00%~86.00%),CD3+CD8+15.00%(正常值范围:13.00%~39.00%),CD3+CD4+49.00%(正常值范围:33.00%~58.00%),B淋巴细胞20.00%(正常值范围:5.00%~22.00%),自然杀伤细胞(NK)细胞14.00%(正常值范围:5.00%~26.00%)。2月25日龄时再次因呛咳20余天,伴气促、咳嗽10余天入院。复查淋巴细胞亚群示:CD3+:38.90%(正常值范围:56.00%~86.00%),CD3+CD8+11.72%(正常值范围:13.00%~39.00%),CD3+CD4+26.82%(正常值范围:33.00%~58.00%),B淋巴细胞53.24%(正常值范围:5.00%~22.00%),NK细胞4.35%(正常值范围:5.00%~26.00%);复查PTH 0 min45.30 pg/ml(正常值范围:18.50~88.00 pg/ml),复查钙离子1.82 mmol/L。⑤病例5,男,因发现CHD43天,气促伴发绀7天入院。患儿系33+6周出生,出生后第6天外院心脏彩色超声发现VSD并ASD,入院查体见小下颌和高腭弓,心脏彩色超声考虑VSD并ASD,肺部超声未探及胸腺,IgG:6.41 g/L(正常值范围:8.60~17.40 g/L),IgA:0.07 g/L(正常值范围:1.00~4.20 g/L),IgM:0.23 g/L(正常值范围:0.50~2.80 g/L)。电解质无异常。11月龄时于中南大学湘雅二医院儿童医学中心小儿心血管外科予VSD修补术及ASD修补术。易罹患感冒,生长发育落后。见表1。
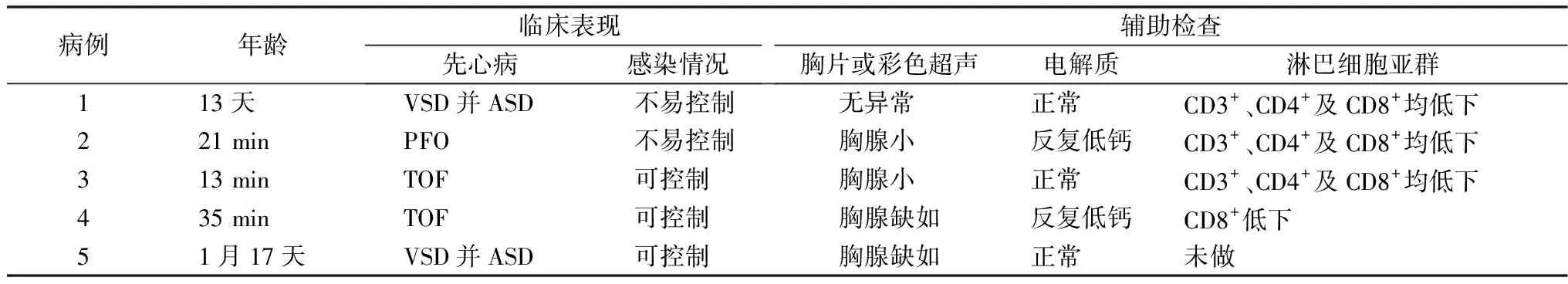
表1 5例临床表现及辅助检查
2.3基因检测 5例中新生变异4例,遗传自母亲1例。缺失片段大于2.45 Mb的3例,缺失片段估测3.0Mb左右(标本送检国内两家医学检验公司)的2例。其中一家医学检验公司在缺失大于2.45 Mb区域缺失基因包括ABHD17AP4、AIFM3、ARVCF、BCRP5、C22orf39、CA15P1、CCDC188、CCDC74BP1、CDC45、CLDN5、CLTCL1、COMT、CRKL、DGCR11、DGCR2、DGCR5、DGCR6、DGCR6L、DGCR8、ESS2、FAM230G、FAM246C、GNB1L、GP1BB、GSC2、HIRA、IGLL4P、KLHL22、KRT18P5、KRT18P62、LINC00895、LINC00896、LINC01311、LINC01637、LINC02891、LZTR1、MED15、MIR1286、MIR1306、MIR185、MIR3618、MIR4761、MIR6816、MRPL40、PI4KA、POM121L4P、PRODH、RANBP1、RN7SL168P、RN7SL812P、RNU6-225P、RNY1P9、RTL10、RTN4R、SCARF2、SEPTIN5、SERPIND1、SLC25A1、SLC9A3P2、SMPD4P1、SNAP29、SNORA77B、TANGO2、TBX1、TMEM191A、TRMT2A、TSSK1A、TSSK2、TXNRD2、UFD1、USP41、ZDHHC8及ZNF74等共73个基因。
3 讨 论
22q11.2DS的遗传病理基础是染色体22q11.2区域0.7~3.0 Mb大小的片段缺失,造成缺失的主要原因是22q11.2上的低拷贝重复序列(low copy repeats,LCRS)减数分裂期间出现不对称非等位的同源重组,其中85%的患者为经典3.0 Mb微缺失,60%~70%的经典微缺失患者患有CHD,主要为圆锥动脉干畸形。90%~95%的22q11.2DS病例为新生突变,6%~28%呈常染色体显性遗传,缺失来源于母亲的略多于父亲[3]。
22q11.2区域有8个LCRS,在减数分裂的过程中,这些LCRS在很大程度上是不能区分的,导致非等位基因的同源重组,这8个LCR22横跨22q11.2区域,分别称为LCR22A-LCR22H,和该病相关的3.0 Mb区域中有4个LCR22,分别为LCR22A、LCR22B、LCR22C及LCR22D。大多数患者在LCR22A-D区域发生缺失为LCR22A-D缺失型;LCR22A-B之间的1.5 MB缺失为第二常见的缺失类型,而2.0 MB的LCRA-C缺失的频率最低,患者通常有上述3种常见缺失。目前45个已知蛋白编码基因定位于3.0 Mb大小的22q11.2区域[4],包括DGCR、TBX1和TUPLE在内的重要基因。有研究表明,TBX1基因功能的丧失或获得可能是导致22q11.2DS的主要原因[5],其次TBX1基因的缺失与免疫缺陷有关(免疫缺陷和胸腺发育不全及继发T细胞生成受损有关)[6]。TBX1基因的缺失和22q11.2DS的5个主要表型(异常面容、心脏缺陷、胸腺发育不良、腭裂-腭咽闭合不全、甲状旁腺功能不全与低血症)相关[7]。王云英等[8]报道了产前彩色多普勒超声诊断CHD胎儿36例,检测引产前脐静脉血标本TUPLE基因位点发现缺失17例,缺失率达47%,在复杂性心脏病胎儿中的缺失率达67%,考虑和TUPLE基因变异后的胚胎不能进行轴向旋转和心脏环化而致CHD有关。
基因型与临床表型的相关性目前尚不明确。Cirillo等[9]指出研究基因型与表现型相关性比较困难,因为基因片段缺失的大小与临床症状严重程度无关,较小基因片段缺失与较轻的症状无关。Rozas等[10]对22q11.2DS荟萃分析发现,基因缺失片段的大小与CHD或腭部异常缺乏相关性,基因缺失片段的大小不能解释上述两种主要表型的不完全外显率,并且心脏发育和腭部异常发生在LCR22 A-B这段关键区域内。但Crowley等[11]通过回顾性分析得出以下结论:与嵌套和远端缺失患儿(LCR22 B-D,LCR22 C-D,LCR22 D-E和LCR22 D-F)相比,22q11.2DS近端缺失患儿(LCR22 A-B,LCR22 A-C和LCR22 A-D)的CD3+和CD4+T细胞计数显著降低。
22q11.2DS典型临床特征为心脏圆锥干畸形,胸腺发育不良及低钙血症,但表现的形式差别很大,即使同一个家庭的成员也可有显著的差异[12-13]。非免疫方面的临床表现中,腭异常占69%~100%,语言障碍占9%~90%,学习障碍占45%~90%,心脏畸形占49%~83%,发育迟缓占75%,眼科异常占7%~70%,低钙占17%~60%,精神异常占9%~60%,骨骼异常占17%~45%,肾脏异常占31%~37%,身材矮小占20%,神经系统占8%,牙科占2.5%。而反复感染为晚期死亡的重要原因之一。Campbell等[14]对1 421例22q11.2DS患者进行回顾性研究显示,在CHD中VSD占23%,法洛四联症(TFO)占18%,主动脉弓畸形占14%,主动脉弓离断占11%,ASD占10%,肺动脉闭锁占6%,永存动脉干占4%,动脉导管未闭(PDA)占6%,二叶主动脉瓣占3%,肺动脉狭窄占2%,其他占1%。本研究5例中VSD合并ASD共2例,TFO2例,发育迟缓4例,与既往文献报道一致。
在某种程度上,报道的22q11.2DS患病率取决于患者的年龄。胎儿期间,与22q11.2DS相关的记录最充分的发现是与唇腭裂和(或)宫内生长受限相关的CHD。在新生儿期,低钙血症、CHD、胸腺缺失和鼻咽反流是有用的临床线索。在婴儿期,面部特征变得更加突出,但临床表现主要以运动和语言的里程碑式延迟为特征。相反在青春期,神经精神疾病和(或)学习障碍的患病率显著增加,如少年至成人阶段人群精神疾病的发病率达到60%[9]。而Sullivan等[15]报道新生儿22q11.2DS中低钙血症的发生率高达74%。在本研究5例新生儿中,仅有2例为新生儿低钙血症,远低于文献报道。
随着22q11.2DS患者数量的逐渐增多,对该病疾病谱的认识也逐渐深入,先天性心脏缺陷、语言延迟和免疫缺陷是22q11.2DS最常见的表型[16],但由于新生儿的特点,先天性心脏缺陷及免疫缺陷在新生儿期常见。
22q11.2DS免疫缺陷即可表现为胸腺发育不良造成T细胞缺失,也可表现为T细胞数量正常[17-18]。有75%~80%的22q11.2DS婴儿T细胞数量较低。因缺乏T细胞婴儿在给予同种异体细胞后可能发生移植物抗宿主病,故许多大型中心医院对给予婴儿的所有血液制品进行辐照。而新生儿的淋巴细胞计数应大于2.8×109/L。按胸腺受累程度的不等,分为完全性的和部分性的22q11.2DS,完全性22q11.2DS胸腺完全缺如,CD3+T细胞<1%~2%,表型同重症联合免疫缺陷(severe combined immunodeficiency,SCID),预后极差,常造成早期死亡,而目前SCID在新生儿期可通过T细胞受体删除环(T cell receptor excision circles,TRECs)进行筛查。尽管新生儿22q11.2DS的诊断主要依赖分子细胞遗传学检测和识别与该综合征相关的表型特征,但同样对新生儿进行TRECs筛查,仍可早期识别患有严重T淋巴细胞减少的22q11.2DS新生儿。对加州国家统计局300多万TRECs结果的回顾性研究发现,22q11.2DS导致19%的新生儿出现非SCID的T淋巴细胞减少[19],故通过TRECs筛查有可能检出完全性22q11.2DS新生儿。完全性22q11.2DS需胸腺或完全匹配的T细胞移植,从供体采集胸腺组织并进行培养,去除能导致移植物抗宿主病的成熟T细胞。胸腺薄片植入股四头肌后90~100天功能性T细胞出现,随后植入胸腺退化而较长时间不产生T细胞。虽存在中度残留免疫缺陷,但患儿仍能产生大量T细胞,并有足够的宿主防御能力,可以正常玩耍和上学。而大多数患儿为部分性22q11.2DS,胸腺常发育不全,可随年龄的增加渐恢复至大致的正常水平。胸腺大小几乎不可能测量,因胸腺的小部分可隐藏在颈部或纵隔结构中,影像学上或手术时所见的非常小的胸腺并不一定代表低T细胞计数,故需对免疫系统进行特定的实验室分析。本研究中,除例5外,其他4例均为部分性22q11.2DS。
22q11.2DS诊断标准[20]分为确定诊断、可能诊断及可疑诊断。确定诊断为:男或女CD3+T细胞数目减少(<500×106/L),且具备以下3项特征中的2项:①心脏圆锥动脉干畸形(动脉单干,TFO,主动脉弓离断,或右锁骨下动脉迷走);②低钙血症持续超过3周并需要治疗;③染色体22q11.2缺失。可能诊断:男或女CD3+T细胞数目减少(<1.5×109/L)和染色体22q11.2缺失。可疑诊断:男或女CD3+T细胞数目减少(<1.5×109/L),且具有以下其一:①心脏畸形;②低钙血症持续超过3周并需要治疗;③特殊面容或腭异常。目前遗传学检测方法主要是荧光原位杂交(fluorescence in situ hybridization,FISH),如FISH检测22q11.2微缺失阴性,可行TBX1基因序列测定。但目前FISH已被比较基因组杂交所取代,用于检测整个基因组而不仅仅限于22q11.2区域。其他方法还有多重连接探针扩增及拷贝数变异方法。
随着产前诊断技术的进步,22q11.2DS可通过产前明确诊断,对降低新生儿出生缺陷意义重大。侯磊等[21]对19例产前诊断22q11.2DS的孕妇(19例中引产18例)的研究发现,胎儿超声异常15例,最常见胎儿超声为右位主动脉弓(8/16)、VSD(5/16)及法洛氏四联症(5/16) ,提示胎儿心血管超声异常是产前22q11.2DS主要表现。罗晓辉等[22]对产前基因确诊的22例22q11.2DS病例研究发现,心脏超声结构异常13例,心脏结构异常合并腭裂1例,足内翻4例,肾脏超声异常3例,胎儿发育迟缓并唐氏筛查高风险1例,而足内翻仅次于心脏结构异常,增加了胎儿22q11.2DS表型,本研究中的病例2胎儿期有左肾积水,支持文献报道。陈铎等[23]通过羊水基因检测确诊6例22q11.2DS,存在2.54~3.2 Mb微缺失,其中母源性遗传1例,父源性遗传1例,新生变异4例,其中4例均选择引产。
新生儿期免疫力低下习以为常的传统思维,特殊面容、先天性心脏畸形和低钙血症在新生儿期病因的复杂性,胸片、彩色超声、CT或手术中所见均不能反映胸腺发育不良或缺如均导致22q11.2DS在新生儿期不能早期发现。产前有先天性的心脏畸形、足内翻及肾脏超声异常等而未行产前基因诊断和有CHD、感染不易控制、反复低钙血症、胸腺小或缺如的新生儿均应考虑该病的可能,及时行基因检测以避免出生缺陷及过度治疗。
