儿童川崎病合并轻微脑炎/脑病伴可逆性胼胝体压部病变综合征1例并文献复习
2022-08-08杜燕燕季丽娜徐樨巍
杜燕燕,王 健,贺 兰,季丽娜△,徐樨巍, 2
(1. 清华大学附属北京清华长庚医院儿科,清华大学临床医学院,北京 102218;2.首都医科大学附属北京儿童医院消化内科,国家儿童医学中心,北京 100045)
川崎病(Kawasaki disease)是一种好发于婴幼儿的不明原因的系统性血管炎,最常见的并发症为冠状动脉病变,约发生在15%~25%未经治疗的患儿中,并可能导致缺血性心脏病或猝死[1]。川崎病合并神经系统并发症多见于无菌性脑膜炎、短暂的单侧面神经麻痹等,合并脑病的病例较少,约 0.1%,多为8岁以上的年长儿[2]。近年来,随着磁共振成像(magnetic resonance imaging,MRI)技术的普及和开展,轻微脑炎/脑病伴可逆性胼胝体压部病变综合征(mild encephalitis/encephalopathy with a reversible splenial lesion,MERS)的报道逐渐增多。该病为一种临床-影像学综合征,临床表现为轻度的意识障碍和行为改变等神经系统症状,头颅MRI具有特征性和可逆性改变。目前为止关于川崎病合并MERS的报道非常少见,对其诊治尚缺乏经验。现对北京清华长庚医院儿科2021年6月收治的1例川崎病合并MERS患儿的临床表现及其诊断经过进行总结,并对相关文献进行复习,以提高儿科医师对该病的认识。
1 临床资料
患儿,男,7岁11月,因“发热、咳嗽6 d,发现皮疹5 d,眼红3 d”于2021年6月4日就诊于北京清华长庚医院儿科。患儿曾在外院予头孢类抗生素抗感染治疗4 d,体温仍反复持续在39 ℃以上,咳嗽不重,少痰。发病以来,患儿精神减弱,食欲下降,平素身体健康,无家族遗传史及类似疾病史。
入院时查体:精神稍弱,全身散在大片多形性充血样红斑,部分皮疹中央可见坏死样变,部分有水疱伴血痂;左侧颈部可触及数枚肿大淋巴结;双眼球结膜充血,未见异常分泌物;口唇干红皲裂,舌乳头充血突起;咽充血,扁桃体Ⅰ度肿大,未见分泌物。双肺呼吸音粗,未闻及干湿啰音。心律齐,心音有力,心尖部可闻及2/6级收缩期杂音。腹部及神经系统查体未见异常。手足末端无硬肿脱皮。
实验室检查:血常规:白细胞计数10.39×109/L,中性粒细胞百分比80.4%,血红蛋白113 g/L,血小板193×109/L;C-反应蛋白48.44 mg/L;降钙素原2.06 μg/L;红细胞沉降率16 mm/h;血钾2.96 mmol/L,血钠128.2 mmol/L;高敏肌钙蛋白T 0.051 μg/L;氨基末端B型利钠肽前体3 206 ng/L;凝血功能、肝肾功能大致正常;呼吸道病原学检查和血培养均阴性;自身抗体:抗核抗体(+)(胞浆型1 ∶160),余均阴性;补体C3、C4正常;尿常规(-)。
其他检查:胸部X线片:双肺纹理略增多,模糊;心电图:窦性心律,T波改变,QTc间期延长;心脏超声:二尖瓣少量反流,冠状动脉内径未见明确异常;腹部超声:未见异常。
患儿临床表现符合川崎病诊断标准,入院当天立即予大剂量丙种球蛋白(2 g/kg)静脉滴注及阿司匹林40 mg/(kg·d)口服,同时不除外呼吸道感染,予哌拉西林他唑巴坦静脉滴注抗感染,以及补钾、营养和保护心肌细胞对症治疗。丙种球蛋白静脉滴注24 h后患儿仍有发热,精神反应弱,且出现嗜睡症状,并诉头痛,神经系统查体未见病理征和脑膜刺激征,头颅MRI提示胼胝体压部局限性梭形肿胀,T1稍低、T2稍高异常信号,扩散加权成像(diffusion-weighted images,DWI)呈明显高信号,表观扩散系数(apparent diffusion coefficient,ADC)图呈明显低信号,提示MERS(图1A、B)。同时患儿Kobayashi评分[3]为5分,考虑可能为丙种球蛋白无反应型川崎病,予甲泼尼龙2 mg/(kg·d)静脉滴注,数小时后热退,头痛、嗜睡症状消失。入院第7天川崎病体征基本消退。入院第12天复查头颅MRI提示胼胝体异常信号消失(图1C、D)。激素2周内逐渐减量至停用,第14天出院,复查心电图、血电解质、高敏肌钙蛋白T、氨基末端B型利钠肽前体均正常,心脏超声二尖瓣反流消失。现门诊随访中,出院后1周出现双手指末端膜样脱皮,无任何神经系统异常,监测冠状动脉内径未见异常,复查抗核抗体阴性。
2 分析与讨论
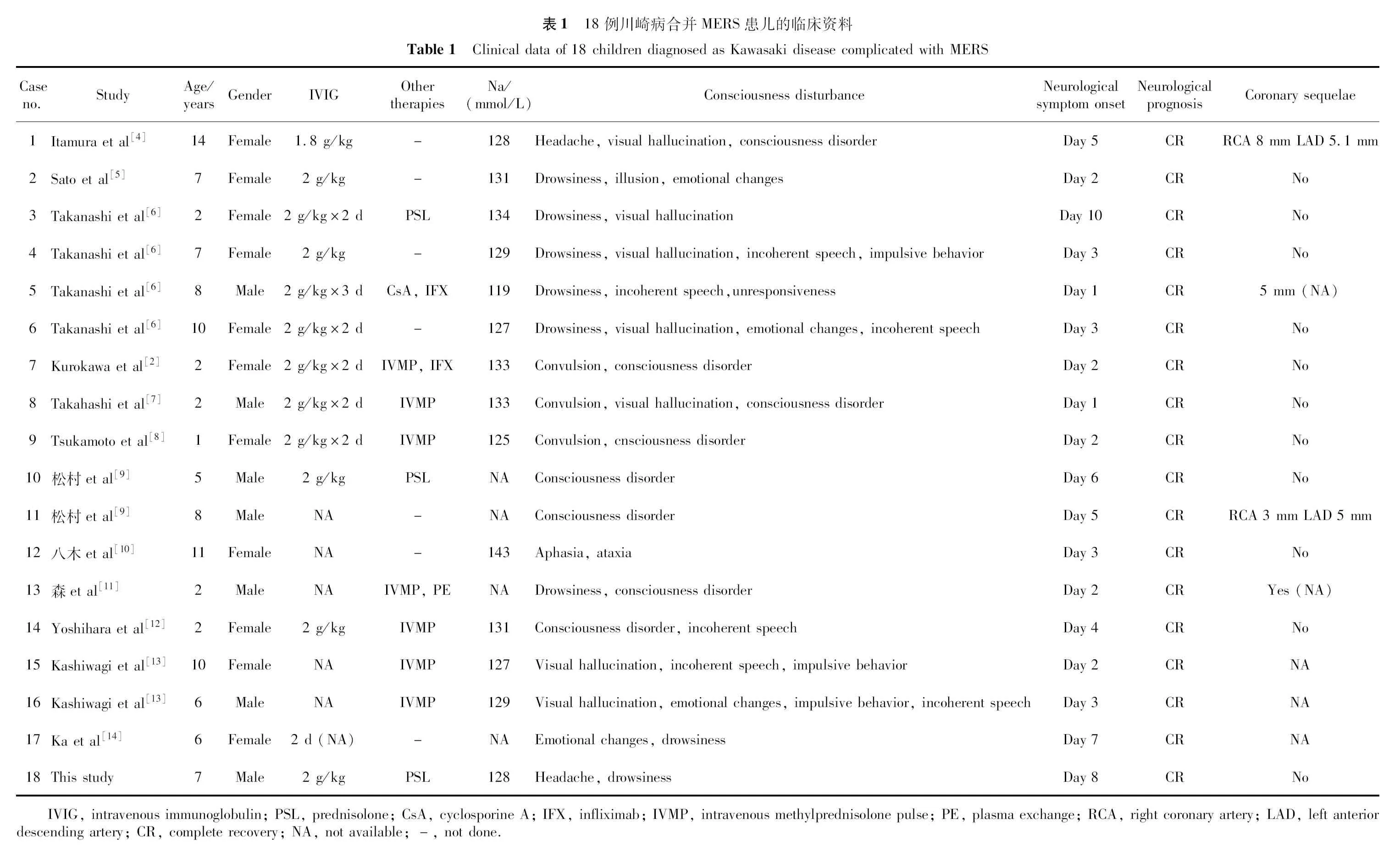
以“川崎病”“轻微脑炎/脑病伴可逆性胼胝体压部病变综合征”“Kawasaki disease”“mild encephalitis/encephalopathy with a reversible splenial lesion”“MERS”为关键词,对万方数据知识服务平台、中国知网(China National Knowledge Infrastructure,CNKI)、维普中文期刊服务平台、PubMed进行文献检索,检索起止时间为建库至2021年6月10日,共检索到符合条件的外文文献12篇[2,4-14],未检索到中文文献。2011年,Itamura等[4]首次报道了1例14岁女童川崎病合并MERS,目前为止共有17例报道,其中16例来自日本,1例来自澳大利亚,我国尚未见类似报道,总结见表1。
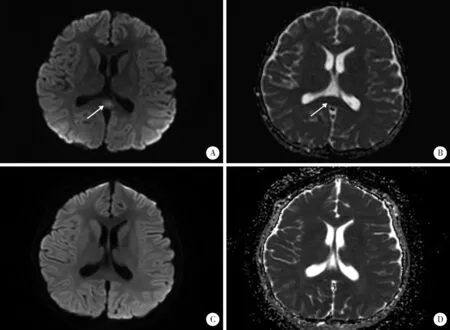
A, diffusion-weighted images (DWI) revealed area of high intensity in the splenium of the corpus callosum (arrow) before treatment; B, apparent diffusion coefficient (ADC) revealed area of low intensity in the splenium of the corpus callosum (arrow) before treatment; C, D showed that the abnormal signal lesions of the splenium of the corpus callosum disappeared after treatment.图1 治疗前后头颅MRI图像Figure 1 Cranial MRI before and after treatment
18例儿童川崎病合并MERS的患儿中最小年龄为1岁10个月,最大年龄为14岁, 其中5岁以上儿童12例(66.7%);女性11例,男性7例。18例患儿中有血钠值报告者14例,按照血钠正常范围135~145 mmol/L,符合低钠血症者13例。所有病例均在川崎病的急性期出现了神经系统症状,表现为意识障碍者15例,幻视及幻觉8例,行为、情绪及言语改变8例,惊厥3例,头痛2例,失语伴共济失调1例。所有病例均在神经系统症状出现后立即行头颅MRI,均提示MERS。经2~6 g/kg大剂量丙种球蛋白及阿司匹林治疗后,7例患儿神经系统症状缓解,11例患儿神经系统症状未改善。这11例患儿中3例经使用小剂量激素治疗(包括本例), 5例经使用甲强龙静脉冲击治疗后病情获得缓解;1例(例5)予英夫利昔单抗联合环孢素治疗后病情缓解;1例(例7)在甲强龙冲击治疗后仍高热不退、意识障碍,加用英夫利昔单抗治疗后上述症状缓解;另1例(例13)予甲强龙冲击治疗后仍发热、意识障碍,同时冠状动脉扩张,予5次血浆置换后病情缓解。其中13例患儿有头颅影像学复查记录,影像学改变均完全消失,所有患儿神经系统症状于1~7 d内逐渐改善,未遗留神经系统后遗症。有冠状动脉病变记录的15例患儿中冠状动脉扩张者4例,发生率为27%。
MERS是一种基于影像学表现的临床综合征,2004年由日本学者Tada等[15]首次提出,2009年Takanashi[16]对其进一步报道。其前驱症状为发热、咳嗽、呕吐、腹泻等,1~7 d后出现脑病表现,常见的神经系统症状包括行为改变、意识改变和癫痫发作,急性期头颅MRI典型表现为胼胝体压部孤立性的T1稍低、T2稍高异常信号,DWI呈明显高信号,ADC呈明显低信号,依据影像学受累部位可分为Ⅰ型和Ⅱ型,两者病理生理过程相同,Ⅰ型病变局限于胼胝体压部,Ⅱ型病变除胼胝体外还有周围白质对称性病变[17]。上述神经系统症状和影像学改变均可在几天到几周的随访过程中完全消失,不遗留神经系统后遗症。
MERS的病因主要为各种传染性疾病,如甲型流感、腮腺炎、麻疹、水痘、腺病毒、轮状病毒、大肠杆菌、沙门菌等,其次为服用抗癫痫药,抗癫痫药中断,体质量急剧下降,代谢紊乱,如低血糖、低钠血症等均可引起MERS。近10年来逐渐有儿童川崎病并发MERS的报道。
MERS的确切发病机制尚不明确,目前认为缺血引起的细胞毒性水肿可能在MERS中起一定的作用,与其他脑区相比,胼胝体神经元、星形胶质细胞和少突胶质细胞的受体密度更高,包括细胞因子受体、谷氨酸受体、毒素受体和药物受体,这种高密度受体导致该部位容易发生细胞毒性水肿,而胼胝体压部神经纤维轴突薄,含水量多,对水电解质紊乱的自我调节能力不足,更易受累[18]。Takanashi等[19]认为MERS与低钠血症相关联,低钠血症可引起脑水肿、髓鞘内水肿及细胞毒性水肿。14例有血钠资料的患儿中低钠血症者13例,占93%,支持低钠血症引起MERS的机制学说。但文献报道45%的川崎病患者可出现一过性低钠血症[20],而出现MERS者仅为极少数,因此单纯以低钠血症解释川崎病患者发生MERS的依据不足。
炎性细胞的局部浸润也可能是MERS的发生机制,而川崎病的急性期伴随着免疫系统的激活,血中白细胞介素(interleukin,IL)-1、IL-6、IL-8,肿瘤坏死因子(tumor necrosis factor,TNF)α以及血管内皮生长因子(vascular endothelial growth factor,VEGF)均高于正常[21]。研究表明,VEGF升高可导致血管渗漏、低蛋白血症和非心源性水肿[22],这些改变可能发展为MERS。同时有学者报道脑脊液中的IL-6升高可能与MERS的发病机制相关[23]。文献报道中只有1例患儿行血IL-6、IL-8、TNF-α、VEGF检测,2例同时行血及脑脊液IL-6、IL-18、TNF-α检测,均明显升高,经对症治疗后复查下降至正常。目前,炎症因子参与MERS的发病机制还需要更多的临床病例进一步研究。
川崎病本身可合并脑血管炎,脑血管炎导致脑灌注压降低也可能参与MERS的发生机制。Ichiyama等[24]证实21例川崎病患者中有6例在急性期行单光子发射计算机断层扫描(single photon-emission computed tomography,SPECT)出现短暂的局部脑低灌注。Hikita等[25]对22例川崎病患者行SPECT检查,发现4例有神经症状的患者和13例无神经症状的患者均出现脑局部灌注不足。Sato等[5]报道1例川崎病合并MERS患儿头颅MRI仅提示胼胝体压部水肿,而急性期行SPECT则显示双侧扣带回、丘脑、基底神经节、脑干和额叶皮质弥漫性低灌注,此为头颅MRI结果正常的区域,这些边缘系统的异常导致幻视、情绪不稳等神经精神症状,提示川崎病脑灌注压降低可能导致MERS,但该例头颅磁共振血管造影(magnetic resonance angiography,MRA)显示正常,似不支持此假说,提示可能有其他的发病机制参与。
川崎病常发生在5岁以下婴幼儿,在10岁或以上儿童中发病率极低,当年长儿患川崎病时容易被忽略和误诊。本文中有冠状动脉记录资料的15例川崎病合并MERS患儿中4例并发冠状动脉病变(27%),明显高于川崎病整体的冠状动脉病变发病率(1%)[26],且出现冠状动脉扩张的4例中3例为8岁以上年长儿,提示患川崎病的年长儿,尤其合并神经系统症状时,有更高的心血管风险,因此需加强心脏超声的监测,高度警惕冠状动脉病变的发生。
所有患儿中5岁以下幼儿有6例,均为丙种球蛋白无反应型川崎病,其中3例以发热、惊厥起病,其后表现出川崎病的临床症状,且病情较重,意识障碍持续时间长(2~7 d),6例患儿均给予积极的丙种球蛋白和激素治疗,部分联合英夫利昔单抗及血浆置换治疗后病情好转,提示年幼儿川崎病合并MERS可能表现出更重的临床经过以及对丙种球蛋白的无反应性,及时行头颅MRI检查有助于早期诊断,积极予激素及英夫利昔单抗等治疗有助于逆转病情和改善预后。
本例川崎病合并MERS应与以下疾病相鉴别:(1)中枢神经性血管炎:中枢神经系统的中小血管免疫炎性病变,多为慢性病程,临床表现多样,可有头痛、癫痫、认知功能下降等,头颅MRI缺乏特异性,病变易累及双侧皮层、皮层下及深部白质,与本例临床表现及影像学改变不符,可以除外。(2)无菌性脑膜炎:川崎病最常见的神经系统合并症,临床可表现为头痛、呕吐、嗜睡、前囟隆起、脑膜刺激征等,为自限性过程,随川崎病的恢复而痊愈,脑脊液淋巴细胞轻度增多,糖及氯化物正常,头颅MRI大多正常,与本例不符。
综上,当川崎病患儿伴有神经系统异常时,尤其是意识障碍、幻视、惊厥等,一定要警惕MERS,应积极完善头颅MRI明确诊断。尽管既往文献报道大部分MERS不需要特殊治疗,神经系统症状和影像学改变可以在短期内可逆性消失,但川崎病患儿合并MERS可表现为病情危重,部分需要积极给予激素或英夫利昔单抗治疗才能逆转病情和改善预后。另外,川崎病合并MERS多见于年长儿,有并发心血管病变的高风险性,更应加强心脏超声的监测。
