调节氧化镉-炭黑界面高效电催化CO2还原生成CO
2022-08-06王丽君詹新雨郝磊端孙振宇
王丽君,李 欣,洪 崧,詹新雨,王 迪,郝磊端,孙振宇
(北京化工大学化学工程学院,有机⁃无机复合材料国家重点实验室,北京 100029)
1 Introduction
The dependance and increasing consumption of finite fossil fuels leads to excessive anthropogenic emissions of CO2and intensifies climate change and energy shortage[1]. To ameliorate these issues,electro⁃chemical CO2reduction(ECR)driven by electricity from intermittent renewable energy sources provides a promising avenue,which also enables a carbon-free economy[2—6]. However,CO2is chemically inert and kinetically stable. Conversion of this C1molecule demands a high energy input for its activation. In addition,the competing hydrogen evolution reaction(HER,from proton and water reduction)concurrently occurs under similar or even lower overpotentials with more rapid kinetics,adversely affecting the ECR selectivity[7]. The pioneering ECR dates back to the 1950s[8]. Subsequent studies by Horiet al.[9]reported four groups of metal electrodes for the ECR in 1985. CO is an essential feedstock for the fabrication of synthetic fuels(>C1hydro⁃carbons or alcohols,light olefins or aromatics)through(modified)Fischer-Tropsch process[10]. Electrochemi⁃cal CO2valorization to yield CO is economically practical based on the high market price and large market size of CO[11]. Noble metals,such as Au[12],Ag[13],and Pd[14,15]are widely used to catalyze the ECR to generate CO because they have medium hydrogen overvoltages and weak CO adsorption properties. However,the scar⁃city and rising cost of the precious metals hamper their large-scale implementation in CO2electrolysis. Hence the design and development of inexpensive and earth-abundant electrocatalysts with high activity,selectivity,and durability for CO2-to-CO conversion are desirable.
In this paper,we demonstrated highly efficient ECR under ambient conditions by tuning the interface of commercial cadmium oxide(CdO)and carbon black(CB). The overall faradaic efficiency(FE)could be attained above 80% within the applied voltage range from −1.0 V to −1.2 V(versusreversible hydrogen electrode,vs. RHE)on the composite catalyst,approaching 92.7% at −1.0 V(vs. RHE),significantly out⁃performing bare CdO catalyst(69.5%). The FE toward CO formation reached 87.4%. Collective knowledge from multiple control experiments manifested that the introduction of conductive CB and the large contact area between CdO and CB contribute to enhanced CO2adsorption and activation,thereby boosting the ECR to yield CO.
2 Experimental
2.1 Materials and Reagents
All the chemicals used in this work were of analytical grade and used without purification. CdO(99%),CB,isopropanol(IPA,≥99.7%)and KHCO3(99.5%)were purchased from Aladdin. Nafion solution(5%,mass fraction),Toray carbon paper and Nafion membranes were provided by Alfa Aesar. Deionized water(DI,18.2 MΩ∙cm)was obtained from a Millipore system. Carbon dioxide gas(99.999%)and argon gas(99.999%)were bought from Beijing Haipu Gas Co.,Ltd.
2.2 Preparation of CdO/CB Composite Catalyst Ink
In a typical procedure to prepare CdO/CB composite catalyst ink,2 mg of CdO and 8 mg of carbon black were dispersed in 240 μL of IPA/H2O(volume ratio=1∶1)and 1.2 μL of 5%Nafion solution to obtain a homo⁃genous suspension,which was thoroughly mixed by ultrasound for 30 min. By manipulating the mass of CdO added,different inks of CdO/CB catalysts with varying CdO mass fraction were obtained.
2.3 Equipment and Characterization
Powder X-ray diffraction(XRD)patterns were recorded on the D/MAX-RC diffractometer operated at 30 kV and 100 mA with CuKαradiation(λ=0.15418 nm)at a scanning rate of 5°/min. X-ray photoelectron spectroscopy(XPS)experiments were performed using a Thermo Scientific ESCALAB 250Xi instrument. The instrument was equipped with an electron flood and a scanning ion gun. The binding energy was corrected for surface charging by taking the C1speak of contaminant carbon as a reference at 284.8 eV. The XPS spectra were all carefully deconvoluted using the Casa XPS software with a Gaussian-Lorentzian product function with similar half peak width used for an equivalent element. Transmission electron microscopy(TEM)was carried out using a JEOL ARM200 microscope at an accelerating voltage of 200 kV. TEM samples were prepared by depositing a droplet of suspension onto a Cu grid coated with a Lacey Carbon film.
2.4 Electrochemical CO2 Reduction Test
For linear sweep voltammograms in Ar- or CO2-saturated 0.1 mol/L KHCO3solution,1 mg of a catalyst(for CdO/CB,the mass of individual CdO and CB added was based on the CdO mass fraction)was dispersed in the mixture of 100 μL of ethanol,100 μL of deionized water,and 100 μL of Nafion solution(1 %,mass fraction). The mixture was then ultrasonicated for 30 min to form a homogeneous ink. Subsequently,7.95 μL of the dispersion ink was loaded onto a glassy carbon electrode and dried under room temperature.
Linear sweep voltammetry was conducted in 0.1 mol/L KHCO3solution with the CHI 760E(Shanghai CHI instruments Co.,Ltd.,China)electrochemical workstation at a scan rate of 2 mV/s. An Ag/AgCl was used as a reference electrode,Pt wire as a counter electrode,and glassy carbon as a working electrode.Rotating disk electrode(RDE)experiments were run on an AFMSRCE RDE control system(Pine Inc.,USA).Before the experiment,the electrolyte solution in the working compartment was purged with Ar or CO2over 30 min to reach a saturated state. The electrochemical impedance spectroscopy(EIS)experiments were operated in Ar-saturated 0.1 mol/L KHCO3solution at an open circuit potential with frequencies from 106Hz to 10 Hz and amplitude of 5 mV.
For H-type cell tests,1.2 mg of a catalyst was dispersed in 240 μL of a mixture of IPA,DI,and Nafion solution(5%,mass fraction)with a corresponding volume ratio of 120∶120∶1.2 under bath ultrasonication for 30 min to form a homogeneous suspension. The suspension was then loaded onto a Toray carbon paper working electrode with an area of 1.2 cm×1 cm and dried under ambient conditions.
All potentials(E,V)in this study were measured against the Ag/AgCl reference electrode(in saturated KCl solution)and converted to the RHE reference scale by the following equation:
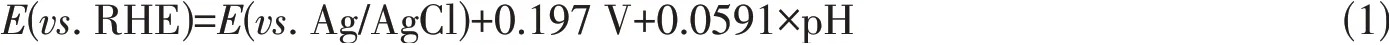
Controlled CO2electrolysis was conducted in an H-cell system separated by a Nafion 117 membrane at room temperature and atmospheric pressure. The cathodic electrolyte was CO2-saturated 0.1 mol/L KHCO3aqueous solution unless stated otherwise and anodic electrolyte was 0.1 mol/L H2SO4degassed under argon.CO2was purged into the 0.1 mol/L KHCO3solution for over 30 min to remove residual air in the reservoir,then controlled potential electrolysis was performed at each potential for 60 min. Prior to the electrochemical measurements,the Nafion membrane was pre-treated by heating in H2O2solution(5%,mass fraction)and H2SO4(0.5 mol/L)at 80 ℃for 1 h,respectively. Subsequently,the treated Nafion membrane was immersed in DI water for 30 min and then washed with DI water repeatedly.
2.5 Product Analysis and Calculations of FE,Partial Current Density,and Production Rate
The ECR gas-phase products were analyzed using an Agilent 7890B gas chromatography(GC)with two thermal conductivity detectors(TCD)and one flame ionization detector(FID). The liquid products were examined by1H NMR(nuclear magnetic resonance,Bruker Avance III 400 HD spectrometer)using a solvent presaturation technique to suppress the water peak. NMR samples were prepared by mixing 0.5 mL of the product-containing electrolyte and 0.1 mL DMSO-d6as the internal standard. FE was determined from the amount of charge passed to produce each product divided by the total amount of charge passed at a specific time or during the overall run. The FE was calculated by the equation as below:

whereZis the number of electrons transferred(Z=2 for CO,HCOOH,and H2production),n(mol)is the number of moles for a given product,F(96,485 C/mol)is Faraday’s constant,Qtotal(C)is all the charge passed throughout the electrolysis process.
Partial current density for ECR products or H2can be obtained by multiplying corresponding FE by the total current density(J,mA/cm2):

The production rate(PR,μmol·of a product was calculated by

whereI(μA)is the total current of all products,m(mg)is the catalyst mass.
The cathodic energy efficiency(EE)for the ECR toward CO was calculated using the following equation[16]:

3 Results and Discussion
3.1 Structural and Morphological Characterization
XRD measurements were performed for bare CdO and the CdO/CB composite. As shown in Fig.1(A),the diffraction peaks at 33°,38.3°,55.3°,65.9°,69.3°and 82°can be well assigned to CdO(JCPDS No.01-075-0592),suggesting a pure phase of CdO[17]without CdO2[18]. After forming composite with CB,the CdO/CB displayed the characteristic peaks of both CB and CdO. XPS investigation was carried out to probe the surface composition of CdO/CB and the oxidation state of the elements. The existence of Cd,O and C was proved by the corresponding wide scan spectrum[Fig.1(B)]. The core level spectra of Cd3dclearly exhibited the Cd3d3/2and Cd3d5/2with biding energies located at 413 and 406.3 eV[Fig. 1(C)],indicating the bonding state of Cd2+,which is in accordance with the XRD result. The two O1speaks centered at 531.5 and 533.1 eV are attributed to Cd—O and C—O bond[8],respectively[Fig.1(D)].
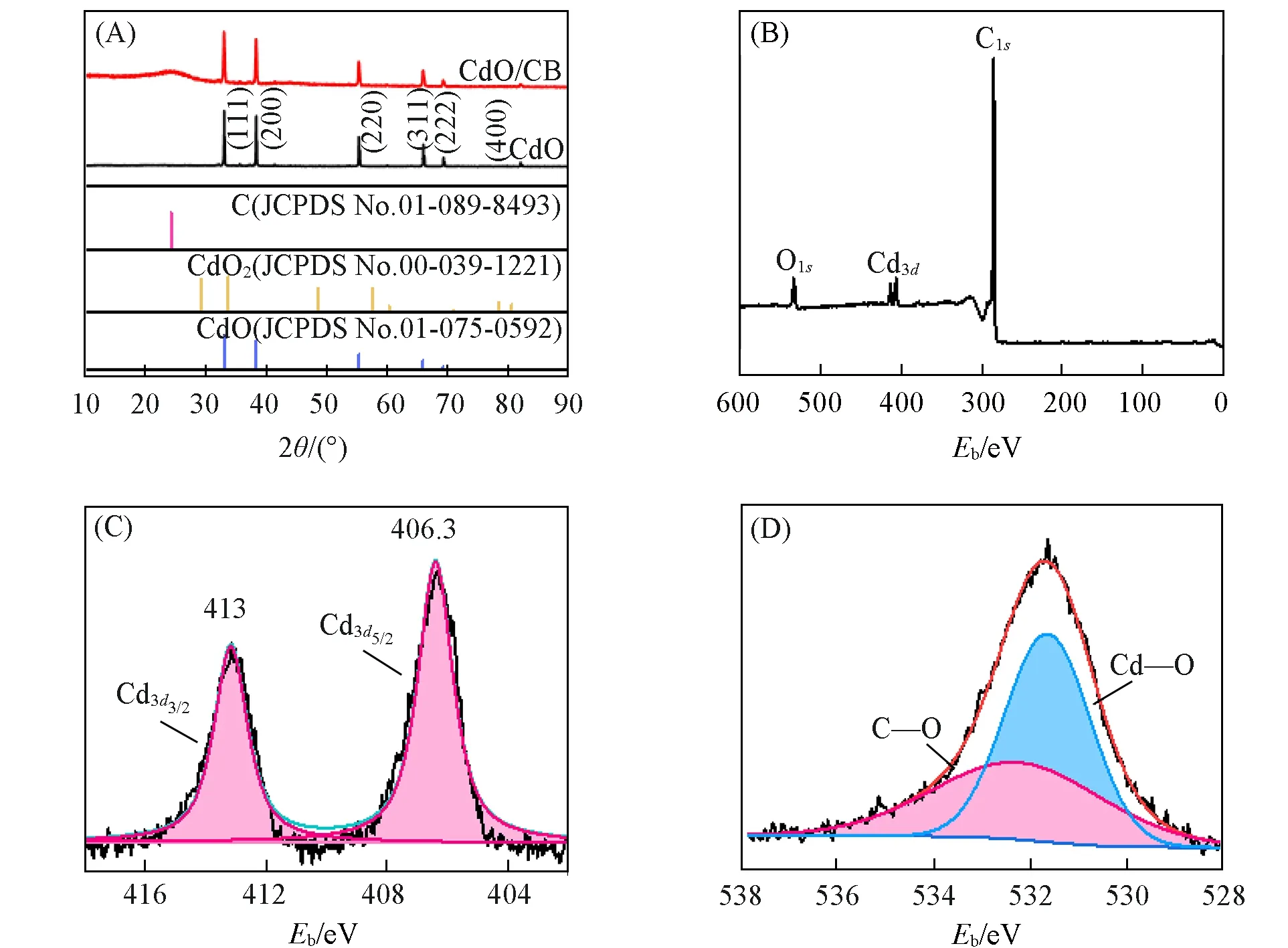
Fig.1 XRD patterns of CdO and 20%CdO/CB(A), wide⁃scan(B), Cd3d(C) and O1s(D) XPS spectra of 20%CdO/CB
The morphology of the CdO/CB composite was examined using TEM and high-resolution TEM(HRTEM).The cubic Monteponite CdO structure was observed from the TEM image of CdO/CB[Fig. 2(A)and(B)].Energy dispersive X-ray spectroscopy(EDS)elemental maps[Fig.2(C—E)]showed the uniform distribution of O,Cd,and C in the CdO/CB composite. Furthermore,the interface between CdO and CB can be identified[Fig. 2(F)]. From the HRTEM image[Fig. 2(G)],the lattice space was measured as 0.27 nm,in good agreement with the lattice parameter of CdO.

Fig.2 TEM image of 20%CdO/CB(A), TEM image of CdO(B) and corresponding EDS elemental maps of C(C), O(D) and Cd(E), HRTEM image of 20%CdO/CB, showing the interface between CdO and CB(F)and HRTEM image of CdO(G)
3.2 Electrochemical Measurements
The ECR catalytic activities of CdO before and after incorporation of CB were investigated. The ECR tests were conducted in a CO2-saturated 0.1 mol/L KHCO3solution(bulk pH=6.8)using a gas tight H-cell separated by a cation-exchange membrane under continuous CO2bubbling[19,20]. Notably,CdO/CB exhibited remarkably higher reduction currents than pure CdO in both Ar- and CO2-purged electrolytes[Fig. 3(A)].This underscores the role of CB in promoting both the HER and ECR. In addition,both CdO and CdO/CB im⁃parted larger current densities in a CO2environment than in an Ar environment over the entire potential range(from −0.4 V to −1.4 V). At potentials <−0.6 V,the cathodic current was observed to increase steeply in CO2-saturated electrolyte,suggesting the predominant occurrence of CO2reduction.
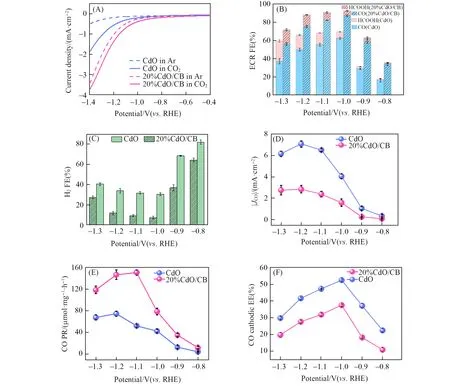
Fig.3 Linear sweep voltammetry(LSV)results of CdO and 20%CdO/CB in Ar or CO2 saturated 0.1 mol/L KHCO3 solution with a scan rate of 5 mV/s(A), ECR FEs(B), H2 FEs(C), and CO partial geometric current densities(D) of CdO and 20%CdO/CB, production rates of CO at different potentials over CdO and 20%CdO/CB(E)and CO cathodic energy efficiency(EE)of CdO and 20%CdO/CB(F)
The reduction products were probed by GC and1H NMR. No ECR compounds were identified in Arpurged 0.1 mol/L KHCO3,indicating that the ECR products were resulted from dissolved CO2from the feed gas. In contrast to pure carbon paper electrode and CB(Fig.S1,see the Supporting Information of this paper)that both predominantly generated H2,both CdO and CdO/CB produced CO and HCOOH along with H2from−0.8 V to −1.3 V,displaying a volcano correlation of FE with switching potential. The total ECR FE and the FE toward CO increased steadily at potentials ranging from −0.8 V to −1.0 V,but dropped when further elevated the overpotential,likely owing to the more intense competition from the HER. Throughout the poten⁃tial regime,the main ECR product is CO. At the scanned potentials below −0.9 V,the ECR dominated over the HER with the overall ECR FE as high as 92.7%at −1.0 V[Fig.3(B)]. Note that the FE of H2evolution decreased across the potential ranges after introduction of carbon black[Fig.3(C)]. The total ECR FE main⁃tained over 88%within the potential range from −1.0 V to −1.2 V. Especially,the maximal FE toward CO for⁃mation on CdO/CB reached 87.4% at −1.0 V(1.4 times that of neat CdO). In addition,higher CO partial geometric current density was attained on CdO/CB over the entire applied voltage range,approaching a maxi⁃mum of 7.1 mA/cm2(2.5 times that of bare CdO)[Fig.3(D)]. Likewise,the CO production rate of CdO/CB is markedly higher than that of CdO throughout the applied bias[Fig.3(E)]. Meanwhile,the energy conver⁃sion efficiency of CdO/CB was calculated to approach 52.5%,in contrast with that of 37.4% for commercial CdO[Fig.3(F)]. It is evident that the CdO/CB invariably outperforms pure CdO in terms of total ECR FE,CO FE,CO partial current density,CO production rate,and CO cathodic energy efficiency.
The ECR performance could be readily modulated by changing the mass fraction of CB. It is worth noting that a dramatic increase in CO partial current density based on CdO mass was achieved upon addition of CB before reaching a peak value of about 32.5 mA/mgCdOin stark contrast to 0.8 mA/mgCdOfor pure CdO. The optimal CB fraction was found to be 90%. Further increasing the content of CB caused a monotonic decrease of CO partial current density[Fig.4(A)]. This is plausible given the decrease in the number of CdO active sites with an increase in the loading of CB. Based on the metrics of overall ECR FE and CO FE,the optimal dosage of CB is 80%[Fig.4(B)]. It is apparent that the introduction of CB largely inhibited the competing HER and also improved the CO FE,mostly resulting from enhanced CO2adsorption(due to the porous structure of CB)and electrical conductivity.
To examine the significance of the CdO-CB interface,we attempted to regulate the interfacial structure by performing four control experiments. We first compared the ECR performance of the two systems of CdO and CB mixtures. One is formed by shaking the CdO dispersion added with CB during the preparation of electrode films. The other one is formed under ultrasonication. It was observed that the physically mixed CdO and CB without ultrasonication provided a substantially inferior ECR activity compared to the system formed under ultrasonication[Fig.4(C)]. We also altered the cascade deposition sequence of equivalent amounts of CB and CdO during the preparation of electrode filmsviaultrasonication. In both cases(i.e.,first deposition of CdO followed by CB named as CdO+CB;first deposition of CB followed by CdO named as CB+CdO),the obtained ECR performances were lower than that of the CdO/CB which possesses the largest accessible contact interface areas for ECR[Fig. 4(C)]. This points to the possibility that the CdO-CB interfaces with exposed cadmium sites plays a crucial role in facilitating the ECR turnover frequency.
To gain insight into the enhanced activity of CdO/CB,the Tafel plot and electrochemical impedance were analyzed. The Tafel slope of CdO/CB was calculated to be 174.8 mV/dec[Fig. 4(D)],smaller than that of pure CdO(198.4 mV/dec),signifying the more rapid kinetics for ECR over the hybrid catalyst. The rate determining step for CO2reduction was thus derived to be the initial step of the CO•2−generation[21]. Nyquist plot analysis[Fig. 4(E)]showed dramatically smaller charge transfer resistance for CdO/CB compared with pure CdO,mirroring its faster interfacial charge transfer between the working electrode and reactants in the electrolyte to accelerate the CO2conversion[22]. In addition,CdO/CB exhibited a strikingly larger electrochemi⁃cal active surface area(derived from measurement of double layer capacitance)than pure CdO(Fig. S2,see the Supporting Information of this paper),greatly benefiting the ECR. The durability of ECR activity over CdO/CB was evaluated by chronoamperometry measurements. No appreciable loss in CO FE and current density took place even after 10 h of continuous CO2electrolysis,manifesting the good catalytic stability of CdO/CB[Fig.4(F)].
4 Conclusions
The CdO/CB composites with tunable interface were prepared through simple ultrasound sonication. The as-obtained CdO/CB can be used as efficient electrocatalysts for ECR to produce CO. The performance of CdO/CB was influenced by the amount of CB added. With an optimal CdO mass fraction of 20%,a highest overall FE of 92.7%toward ECR was achieved at −1.0 V(vs. RHE)over CdO/CB,much higher than that of bare CdO. Further study indicated that the interface and large contact area between CdO and CB contribute to the enhanced performance of the composites. In light of the low cost and abundance of commercial CdO and CB,this work offers a facile method for manufacturing economic electrocatalysts for efficient ECR.
The supporting information of this paper see http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220317.
