山羊5个组织的全长转录组构建与分析
2022-08-04魏文景孙学良占思远张国俊张红平郭家中
魏文景,孙学良,占思远,陈 瑜,张国俊,苗 斌,张红平*,郭家中*
(1.四川农业大学动物科技学院,成都 611130;2.巴中市南江黄羊科学研究所,四川 南江 635600)
受益于高通量测序技术的迅速发展,目前主要畜禽物种全基因组序列均已发布(http://asia.ensembl.org/index.html)[1];尽管如此,不断完善各物种全基因组序列的注释才能为相关物种重要经济性状遗传分析、基因组选择等研究提供坚实的数据基础。伴随着 Hi-C[2]、CHIP-seq[3]和 RNA-seq[4]等功能基因组学技术的发展,人们得以在基因表达的不同水平上深入注释畜禽全基因组序列,尤其是各种调控元件[5-7]。例如,与Illumina测序平台为代表的短读段测序技术相比,PacBio和Nanopore平台实现了高通量长读段测序,在转录水平上能够捕获全长转录本[8-10],从而更准确地探究可变剪接[11-12]、多聚腺苷酸化[13-14]等问题。
山羊是重要的家畜物种之一,山羊遗传育种的深入研究依赖于完善的山羊基因组序列及注释。目前被广泛使用的山羊参考基因组(ARS1)具有较高组装质量(缺口总长度38 Mb,contig N50值为26.2 Mb等特点)[15];但功能注释并不完善,这些未注释的大量基因可能具有重要的功能。例如,本课题组利用比较基因组学分析,初步确定山羊7号染色体45.05~59.76 Mb区域是影响山羊小耳表型的一个重要位点,但该区域内的大部分序列缺少注释,从而阻碍了因果突变鉴定的深入开展[16]。另一方面,已报道的山羊不同发育阶段、不同组织的转录组研究主要使用短读段测序技术,侧重于基因差异表达分析。例如,Zhan S.Y.对不同阶段山羊胎儿背最长肌组织的RNA-seq进行转录组图谱分析,以识别差异表达基因[17];C.Muriuki根据表达模式将预测转录本分组,并进行聚类分析以完善山羊基因表达图谱数据集[18]。而有关山羊基因功能注释方面的研究偏少[17-20]。本研究收集了南江黄羊的5种组织,利用PacBio测序技术开展全长转录组测序和分析,旨在完善功能注释,从而为山羊遗传育种研究提供理论参考。
1 材料和方法
1.1 组织样品收集
在四川省南江黄羊原种场,选择1只约7月龄健康的雄性断奶山羊。在禁食12 h后进行屠宰,分别收集羊的肝、脾、肾周脂肪、背最长肌和睾丸等组织样品,随后立即置于液氮中,带回实验室保存于-80℃冰箱中。
1.2 测序文库构建和高通量测序
选择肝、脾、脂肪、肌肉和睾丸共5个组织的样品送至北京诺禾致源科技股份有限公司,使用Trizol试剂进行总RNA提取。待RNA样品检测合格后,将上述5个组织总RNA进行等量混匀,形成1个总RNA混合样本。按照PacBio测序说明书,利用带有Oligo(dT)的磁珠富集总RNA混合样本中的mRNA后,进行大规模PCR扩增并进行各种修复,最终获得SMRT bell文库。使用PacBio Sequel测序仪进行高通量测序。
1.3 序列质控和全长转录本序列的鉴定
在获得原始测序数据(polymerase reads)后,使用PacBio官方软件SMRTlink(v.7.0)去除接头序列并过滤掉长度小于等于50 bp的序列,获得子序列(subreads)。对subreads进行校正纠错后,获得环形一致性序列(circular consensus sequence,CCS);再根据CCS是否包含5′端引物、3′端引物以及polyA序列分为全长非嵌合序列(full-length non-chimeric reads,FLNC)与非全长非嵌合序列(non-full-length reads,NFL);然后利用同种类型聚类(ICE)对全长序列进行聚类,获得Cluster consensus序列;最后使用非全长序列对获得的一致序列进行校正;最终获得高质量一致性全长序列(polished consensus reads,PC),被用于后续分析。
1.4 生物信息学分析
使用GMAP[21](v.2017-06-20)软件(默认参数),将高质量一致性全长序列比对到山羊参考基因 组(ARS1,GCA_001704415.1)。 然 后 ,使 用TAPIS[12,22](v.1.2.1)软件对比对上基因组的PC序列进行进一步的校正、聚类去冗余得到转录本。利用GMAP软件对FLNC序列进行饱和度曲线分析,评估测序数据量。将转录本与山羊基因组比对,得到比对上的转录本与未比对的转录本。使用SUPPA[23](v.2.3)软件对比对上的转录本进行可变剪接事件鉴定;该软件将可变剪接分为7类:外显子跳跃(skipped exon,SE)、外显子互斥 (mutually exclusive exon,MX)、5′端 可 变 剪 接 (alternative 5′splice site,A5)、3′端可变剪接 (alternative 3′splice site,A3)、内含子滞留 (retained intron,RI)、起始外显子可变剪接(alternative first exon,AF)、终止外显子可变剪接(alternative last exon,AL)。使用KEGG公共数据库,对比对上与未比对到基因组的转录本序列进行功能注释。
2 结果与分析
2.1 全长转录本的鉴定和比对
对原始序列进行质控,共获得21 078 721个subreads(36.97 Gb),subreads平均长度为 1 755 bp(N50=2 107 bp)。根据全长序列的判定标准,进一步获得341 023条FLNC序列(包含5′引物、3′引物和polyA尾巴),平均长度为1 940 bp(N50=2 242 bp)。对FLNC序列进行校正及聚类去冗余,最终获得28 432条PC序列,平均长度为2 045 bp(N50=2 398 bp)(表1)。上述PC序列被认为是潜在的转录本,被用于后续的基因组比对。
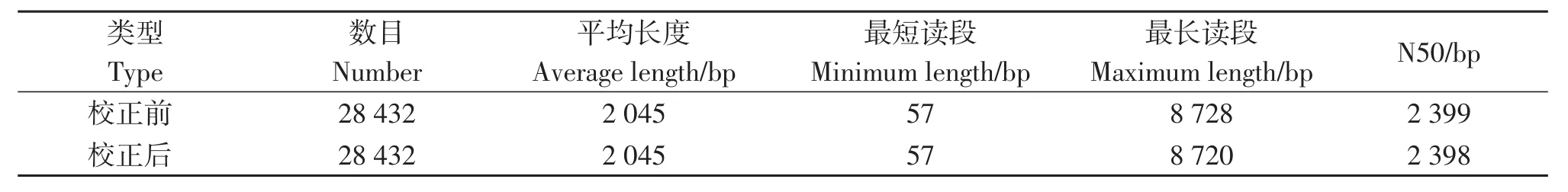
表1 校正前后的转录本长度分布统计表Table 1 The summary of the length distribution of transcripts before and after correction
基因组比对结果表明,99.49%的PC序列(28 288条)被成功比对到山羊基因组,仅有144条序列未比对上(图1A)。在比对上的序列中,93.87%的序列(26 689条)比对到基因组的唯一位置——13 532条序列(47.59%)比对到基因组“+”链,13 157条序列(46.28%)比对到基因组“-”链。另外,饱和度曲线表明,本研究获得的全长序列能全面涵盖相关组织表达的转录本(图1B)。

图1 PC序列的比对结果总结(A)和全长非嵌合序列的饱和度曲线(B)Figure 1 Mapping summary of PC sequences(A)and Saturation curve for full-length non chimera reads(B)
2.2 转录本可变剪接及新基因功能注释分析
对成功比对到基因组的序列进行聚类、去冗余,共鉴定出来源于9 879个基因的19 115个转录本,平均长度为2 069 bp(N50=2 452 bp)。发生转录的基因数量占山羊参考基因组(ARS1)注释基因总数(27 271个)的36.23%。根据基因组注释,9 879个被表达基因包括8 672个已知基因和1 207个新基因。在上述新基因中,1 120个基因位于常染色体上;其中18(65)、19(64)、3(60)和1号(60)染色体上数量较多;其余87个新基因位于Scaffold上;另外,这些新基因总共表达1 790个转录本。
统计分析结果表明,每个基因平均转录形成1.93个转录本(中位数为2个);表达多个(≥2)转录本的基因(4 019)占被转录总基因数的40.68%,如ODF2、BAG6、ALDOA等基因在内的114个基因形成的转录本数目大于或等于10个(表2)。由表3可知,共有4 089个基因发生了可变剪接,占表达基因总数的41.39%。在9 721个可变剪接事件中,AF类型占比最高(36.47%),其次是SE类型的可变剪接(25.44%),最少的是MX类型的可变剪接(1.78%)。

表2 转录本种类和可变剪接事件较多的5种基因Table 2 Five genes with more transcript types and alternative splicing events
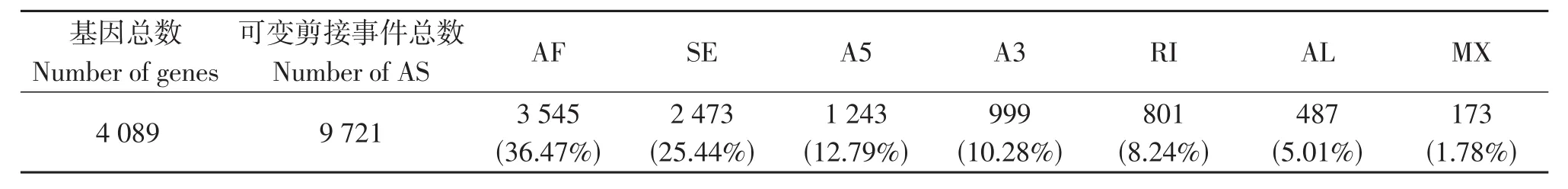
表3 可变剪接事件统计结果Table 3 Alternative splicing events statistics results
功能注释结果表明,在1 207个新基因中共有577个被成功注释到KEGG数据库(图2A)。按照生物体系统(organismal systems)分类,其中23个与免疫系统相关,22个与消化系统相关,19个与内分泌系统相关。依据人类疾病(human disease)分类,与各类癌症相关的转录本共19个;与内分泌和代谢相关的转录本共26个,其中注释到转录本数目最多的基因为GP1BA。上述结果表明,相较于其他功能或表型相关基因,当前的山羊参考基因组中关于免疫和内分泌系统的注释相对较少。
2.3 未比对转录本功能注释分析
由图3A可知,144个未比对到基因组的转录本长度的最小值为388 bp、最大值为6 083 bp、平均长度值为2 186 bp(中位数为1 884 bp)。GC含量分析结果表明(图3B),这些序列GC含量最低值、最高值分别为36.17%、67.86%,平均值为56.50%。对未比对转录本序列与比对上转录本序列两者进行CG含量分析,得到未比对转录本GC含量显著高于比对上的其他转录本这一结果(Wilcoxon秩和检验,P-value=1.12-13)。功能注释结果表明,共有141个转录本序列被成功注释到KEGG数据库(图2B)。按照生物体系统(organismal systems)分类,其中28个与免疫系统相关。依据人类疾病(human disease)分类,与病毒性、细菌性和寄生虫性传染病例相关的转录本共79个,与免疫疾病相关的转录本共26个,其中GC含量最高(67.86%)的转录本被注释到了NUDT14基因。
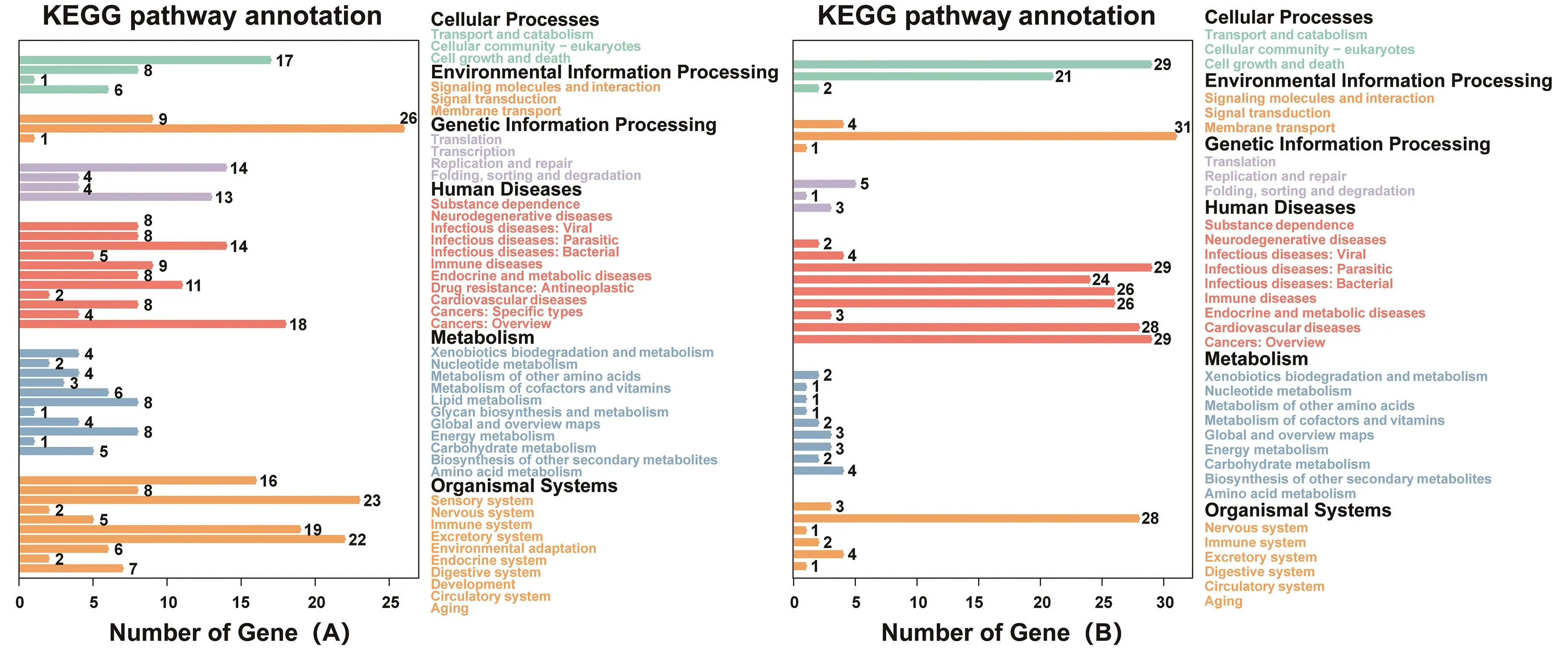
图2 新基因转录本(A)和未比对转录本(B)的功能注释Figure 2 Functional annotations for the transcripts of novel genes(A)and the unmapped transcripts(B)
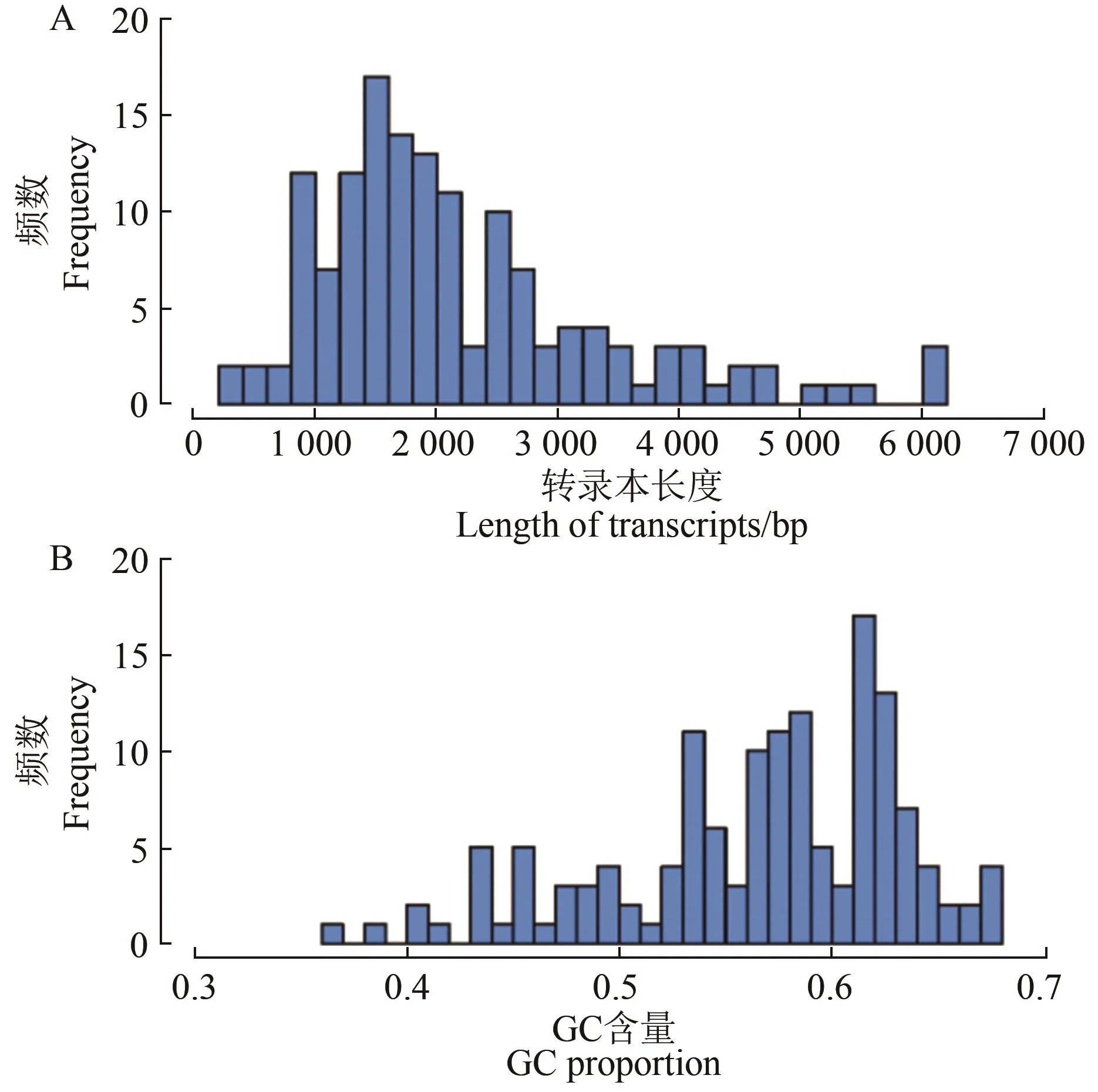
图3 未成功比对到基因组的转录本的长度(A)和GC含量(B)Figure 3 Length(A)and GC content(B)for unmapped transcripts
3 讨论
山羊遗传育种的深入研究依赖于完善的山羊基因组序列及注释信息。目前,“黄金级”高质量山羊基因组(ARS1)[15,24]被广泛使用,但山羊基因组序列的功能注释相对滞后[25]。本研究使用PacBio测序技术对山羊5个组织的混合样本进行转录组测序与分析。饱和度曲线分析结果表明,本研究获得的测序数据全面地涵盖了混合样本中所表达的转录本。在约3万个预测转录本中,超过60%是参考基因的新亚型,该结果与其他家畜物种以及人类基因组的注释结果相似[26],表明山羊基因组转录的复杂性。
本研究发现接近50%的基因转录出多个(≥2)转录本,表明了可变剪接在山羊基因表达过程中广泛存在。已有研究表明,在动物体内SE和AF两种可变剪接类型事件占比最高[27-29],这与本研究结果一致。总的来说,本研究结果显著提高了每个基因平均表达的转录本数量(从1.53个增加到1.93个),这与在水牛中观察到的相近(每个注释基因表达1.91个转录本)[30]。但基于海福特牛32个组织全长转录本研究表明,被转录基因平均表达3.57个转录本[26]。造成上述结果差异主要原因是海福特牛研究包含的组织数量更多,种类更全。另一个原因是上述研究使用了不同的全长测序平台;有研究发现,与PacBio平台相比ONT平台在转录本预测方面假阳性较高、可重复性相对较差[31-33]。
山羊的免疫基因是其抗病能力的遗传学基础;但由于免疫基因复杂的结构以及测序技术的限制,目前相关研究仅在抗寄生虫感染、血液免疫等部分领域有所进展[34-35]。相较于蛋白质编码基因、rRNA、miRNA等其他类型的基因,目前山羊参考基因组关于免疫系统注释相对匮乏。已有研究表明,较高的GC含量是导致免疫基因难以被鉴定的主要原因之一[36]。将脾脏组织加入混样,旨在完善抗性基因相关注释,最终在新基因转录本中鉴定到GP1BA等免疫相关基因。在未比对转录本功能注释中,鉴定到免疫相关转录本数量最多,且GC含量显著高于其他转录本。其中GC含量最高(67.86%)的转录本被注释到了NUDT14基因(已被证明编码蛋白影响病毒DNA复制[37])。免疫基因Ig重链与轻链特殊的组合方式决定了浆细胞产生抗体的特异性;在该机制下,B细胞基因理论重排结果超过2×106个。而目前山羊参考基因组中IgV区段与IgC区段注释共10个[38],远小于理论值。综上,本研究提高了山羊每个基因平均表达的转录本数量;改善了山羊免疫基因的功能注释。
