波纹龙虾全长转录组测序分析
2022-08-04梁妃爽梁华芳孙榕泽徐思行温崇庆广东海洋大学水产学院广东湛江524088
梁妃爽,梁华芳,孙榕泽,徐思行,温崇庆,王 伟 (广东海洋大学水产学院,广东湛江 524088)
转录组(Transcriptome)是指特定细胞或组织中的所有转录产物,包含信使 RNA、核糖体 RNA、转运 RNA以及非编码 RNA。1977年,第1代DNA测序技术(Sanger双脱氧链终止法)开始探索基因结构,但 Sanger 测序方法速度慢、通量低、成本高,难以满足大量测序要求,因此难以应用于组学测序研究。第2代测序技术主要基于Roche/454、ABI/Solid、Illumina/Solexa测序平台,有效解决了速度慢、成本高、通量低的问题,但是其读长短,拼接得到的转录本结构不完整。PacBio平台单分子实时测序技术,也称为SMRT(Single Molecule Real-Time)测序,因其超长的测序读长、超高的测序通量、无GC偏好性、无PCR扩增偏向性、直接检测碱基修饰等诸多优势而广泛应用于水产领域研究,为基因组学、转录组学及DNA甲基化等研究注入了新活力。张金勇等对金乌贼()采用全长转录组测序,筛选SSR位点并分析出现频率较高且类型丰富、多态性潜能较高等特点。Zhang等利用三代测序技术筛选施氏鲟 () 性腺中早期配子发生的相关基因,探讨了其生殖调控机制。Pootakham等利用三代测序技术对斑节对虾生成第1个全长转录组。因此,利用三代测序技术对波纹龙虾全长转录组测序,分析环境因子相关胁迫差异基因,以期为相关功能基因的研究以及波纹龙虾养殖过程中水质调控和抗环境因子变化胁迫等提供理论支持和数据支撑。
波纹龙虾隶属于十足目(Decapoda)龙虾科(Palinuroidea)龙虾属(),是我国沿海地区的重要经济种类,营养丰富,味道鲜美。目前波纹龙虾基因组尚未测序完成,其基因序列信息比较匮乏,相关的分子遗传基础也很薄弱,分子生物学水平研究也较少,仅见李斌等研究了波纹龙虾C-型凝集素PhLecA的基因克隆与表达,Zhuo等研究了波纹龙虾蜕皮激素受体的分子克隆、特征和表达分析,罗嘉俊等研究了波纹龙虾GIH基因克隆、表达及其对光周期的响应等。笔者采用三代测序技术的PacBio 单分子实时测序平台对波纹龙虾()进行全长转录组测序,通过生物信息学方法进行序列拼接、功能注释、分类和代谢通路分析,获取丰富的波纹龙虾序列信息,旨在为进一步挖掘波纹龙虾相关功能基因、基因组学及开发分子标记等研究奠定基础。
1 材料与方法
波纹龙虾为购自海南省琼海市青葛村,体质量为21~57 g的幼龙虾,采用干运法运回广东海洋大学东海岛生物研究基地,在20 m的水泥池中暂养,用黑色幕布遮光,24 h不间断充气。试验海水来源于自然海区,盐度为26‰~30‰,pH 为8.1~8.3,每天换水50%,投喂菲律宾蛤仔()等主要鲜活饵料,养殖21 d后开始试验。取正常条件养殖下波纹龙虾的肝胰腺、鳃、肌肉、性腺、眼柄和80 mg/L亚硝酸盐(NaNO)胁迫7 d的肝胰腺(做3+2项目,取亚硝酸盐胁迫混合作全长转录组,主要是为了进行二代测序时方便筛选差异基因用于后续试验),迅速放入RNAlater中保存,后用干冰保存寄往北京诺禾致源科技股份有限公司进行RNA提取和相关测序。
利用Trizol法分别对建库及定量试验所用的波纹龙虾组织进行RNA提取,使用1%琼脂糖凝胶电泳分析RNA纯度和完整性,Agilent 2100软件准确检测RNA完整性,用Qubit 2.0软件精确定量RNA浓度,Nanodrop软件检测RNA纯度,并选择符合测序标准的RNA等量混合用于文库建设。混合后的RNA样品纯度和完整性:6例混样的Qubit浓度为112 μL,Qubit体积为39 μL,Qubit总量为4.368 μg,样品纯度1.949,1.303,Nano浓度为236.417 ng/μL,RIN值为3.8,NC/QC为2.11。结果表明,该例样本基线平整,符合三代建库标准。波纹龙虾是水产低等动物,RNA只有单峰值,检测时会出现其他峰值干扰从而导致RIN值较低的情况,但6例混样的组织RIN值均符合二代测序要求。
文库的构建流程如图1,构建好的文库进行质量检测后,在Illumina高通量测序平台NovaSeq 6000进行测序(诺禾致源科技有限公司,天津)。

图1 转录组文库构建Fig.1 Transcriptome library construction
测序完成后对原始下机数据进行去接头和低质量reads,得到高质量数据,采用软件SMRTlink v8.0对高质量数据进行序列组装,得到波纹龙虾的转录组基因数据(Unigene)库。序列分析步骤分别是使用下机数据中subreads.bam文件通过CCS算法,对单分子多测序序列进行自我更正,获得CCS (circular consensus sequence)序列;通过检测CCS是否包含poly-A、5′-primer、3′-primer,对CCS进行分类并找出FLNC(full-length non chimera:全长非嵌合序列)序列和nFL(Non-Full-Length:非全长非嵌合序列)序列;将同一转录本的FLNC序列使用hierarchical n*log(n)算法聚类,得到consensus序列;最后对由此产生的全长序列进行polish,获得Polished consensus序列进行后续分析。具体流程见图2。
全长转录组序列的功能注释及结构分析去冗余后的序列使用 CD-HIT 软件进行基因注释,使用的数据库包括:NR, KOG/COG,NT,Pfam,KEGG,Swiss-Prot,GO。
2 结果与分析
转录组测序数据质量分析。波纹龙虾正常条件下的肝胰腺、鳃丝、眼柄、肌肉和性腺,以及亚硝酸盐胁迫下的肝胰腺的转录组测序数据见表1。碱基质量及组成分析显示,GC 含量区间为35.34%~48.26%,各组织样品Q20碱基百分比不小于97.56%,Q30 碱基百分比不小于92.92%。这说明测序产出质量符合要求,能用于后续组装分析。转录本校正分析得出平均序列长度4 147 bp,N50为4 671 bp,注释率为93.74%。

图2 Iso-Seq分析流程Fig.2 Iso-Seq analysis flow chart

表1 数据产出质量情况
CDS预测。CDS(coding sequence)是编码一段蛋白产物的序列。在全长转录组的测序结果中,预测蛋白质编码区有助于基因的初步分析,同时也是进行后续蛋白结构分析的基础。利用ANGEL软件进行CDS预测分析,结果显示共有1 043个基因片段可视为蛋白编码区,其序列长度为0~7 500 bp,主要集中于300~2 500 bp(图3)。

图3 CDS长度分布Fig.3 The statistics of sequence length of CDS
lncRNA分析。LncRNA是一类转录本长度超过200 nt,不编码蛋白质的RNA分子。由于建库原理的限制,只能获得含有polyA尾的lncRNA。使用CNCI、PLEK、CPC2软件以及Pfam数据库对PacBio测序数据进行编码潜能预测,最终分析得到3 105个LncRNA序列,其中共有数目为272个(图4)。
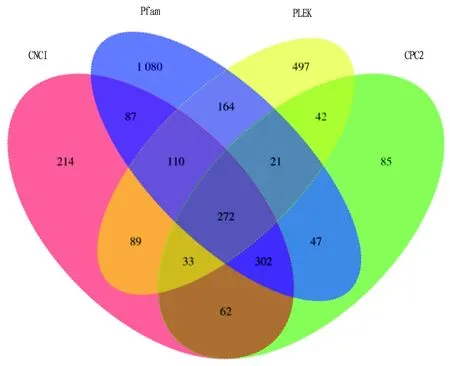
图4 编码潜能预测维恩图Fig.4 Encoding potential prediction Venn diagram
转录本分析。测序结果与数据组装使用PacBio 测序平台对波纹龙虾鳃、肝胰腺、肌肉、性腺和眼柄等组织混样进行全长转录组测序,对原始数据进行过滤,共获得17 044 319个子序列(大小59.47 Gb),平均子序列长度为3 490 bp,N50为4 037 bp。通过每个ZMW孔中子序列的CCS聚类之后得到的序列数为517 682个,序列平均长度为4 181 bp,N50为4 965 bp。同时含有3′引物和5′引物,以及3′引物前含有polyA尾的全长序列(Full-Length,FL)459 737个,全长非嵌合序列(Full-Length non-chimericRead,FLNC)458 653个,序列平均长度为4 094 bp,N50为4 919 bp ,FLNC/CCS为88.60%。全长转录组得到改良后一致序列21 524个,10 425个Unigenes(图5),序列平均长度为4 008 bp,N50为4 474 bp。

图5 波纹龙虾转录本的长度分布Fig.5 Length distribution of Panulirus homarus Unigene
Unigene的功能注释NR数据库注释到Unigene的数量最多,为9 580个,NT数据库注释到的最少,仅3 498个(图6)。

图6 七大数据库注释统计结果Fig.6 Annotation statistical results of seven databases
NR分析。NR数据库注释将波纹龙虾转录组所获得的单基因簇序列在NR数据库中比对,共比对到358个物种,其中钩虾()的同源序列最多,为3 865 个,占注释序列总数的 40.34%,推测波纹龙虾与钩虾同源性较高;其次为湿木白蚁()563个,美洲鲎()291个,大型蚤()282个,凡纳滨对虾 ()171个,斑节对虾()162个,鸭嘴舌形贝()142个,淡水枝角水蚤()134个,中华绒螯蟹()128个,叶蝉()103个,日本囊对虾()96个,白氏文昌鱼()92个,囊舌虫()91个,赤拟谷盗()83个,鞘翅鸟()82个,温室希蛛()74个,裸长角虫兆()72个,淡水螯虾()70个,红螯螯虾()68个,克氏原螯虾()66个,其他物种2 945个。
GO 功能注释。GO 功能注释结果见图7~9,共有7 585条Unigenes被注释分类,从细胞组分可细分为16 类,占比最多的是细胞(cell)和细胞组成(cell part)(44.85%),其次为细胞器(organelle)(17.57%)(图7)。从分子功能细分为10类,其中捆绑(binding)包含Unigenes最多(64.31%),催化活性(catalytic activity)次之(41.08%)(图8)。生物学过程可细分为24 类,细胞过程(cellular process)包含Unigenes最多(42.50%),代谢过程(Metabolic process)类次之(40.07%)(图9)。

图7 细胞组分Fig.7 Cellular component

图8 分子功能Fig.8 Molecular function

图9 生物学过程Fig.9 Biological process
KOG 功能分类。KOG功能注释结果共有7 436条 Unigenes 被注释分类,分布于26 类(图10)。其中只是一般功能预测类共有1 421条注释信息,占比最大 (19.11%),其次为信号转导机制,有1 238条注释信息(16.65%),未知蛋白仅有7条(7.13%)。
KEGG功能注释分析。KEGG功能注释结果显示,共有9 254条 Unigenes 被注释分类,分布于 347 个已知途径中,其中前 12 个代谢途径,注释基因数占总量的 25.26%。前 5 个途径分别是心肌细胞的肾上腺素能信号(ko04261)289 条、病毒性心肌炎(ko05416)254 条、心脏肌肉收缩(ko04260)236 条、癌症中的蛋白多糖(ko05205)200 条和局部黏连(ko04510)191 条(表2)。

表2 前12个代谢途径基因数量Table 2 Number of genes in the first 12 metabolic pathways
转录因子分析。转录因子(transcription factor,TF)作为一类特殊的DNA结合蛋白,可与基因5′末端上游的特定序列结合,使目的基因可以特定时空表达,通过转录因子与其他相关蛋白质的相互作用来激活或抑制转录效果,发挥着重要的调控作用。动物转录因子鉴定使用动物转录因子数据库—animal TFDB 2.0预测到转录因子家族共有543个,属于29个家庭(图11),其中转录因子家族较多的有:zf-C2H2家族有190个、ZBTB家族有110个,TEA家族最少,只有1个,这些转录因子家族成员的获取可为后期波纹龙虾生长发育、代谢调节、免疫应答等相关研究奠定基础。
3 讨论
转录组测序技术是一种成本低,能快速获取大量转录数据并对研究生物体生物学特性、基因功能、相关代谢途径和信号通路等具有重要作用的测序技术。转录组测序分析技术广泛应用于水产养殖相关研究中,并已成为研究环境胁迫对甲壳动物免疫、生长、繁殖以及蜕壳等过程的影响的重要手段之一。

图10 KOG数据库注释统计Fig.10 KOG database annotation statistics

图11 转录因子分析Fig.11 Transcription factor analysis
该研究对波纹龙虾全长转录组进行测序及分析,共获得21 524个转录本,10 425个Unigenes,转录本校正分析得出平均序列长度为4 147 bp,N50为4 671 bp,注释率为93.74%,测序结果可以看出,组装得到序列完整性较好。利用NR、NT、KOG、KEGG等七大公共数据库进行功能注释分类,有9 580 个获得NR数据库注释,对比到358个物种,与钩虾对比的同源信息最多,占40.34%,推测可能是由于钩虾与波纹龙虾的进化史和繁殖习性较为相似。将获得的波纹龙虾单基因簇与GO数据库进行匹配,有7 585个得到GO注释,被划分到BP、CC及MF3大类,涵盖上述功能类别的50个亚类。通过KOG数据库对比波纹龙虾单基因簇,共有7 436个获得注释信息,共分为26个功能组分。与KEGG数据库对比,最终波纹龙虾单基因簇注释到6大类43小类,其中基因数量较多的代谢通路有信号转导机制通路887个。李喜莲等进行红螯螯虾肝脏、卵巢和精巢二代测序获得了6 736条Unigene,注释到GO为16 989个,注释到COG为4 697个,注释到KEGG为9 842个。陈雪峰等对罗氏沼虾卵巢4个不同发育期进行2代测序产生了95 379个Unigenes,注释到GO为6 422个,注释到KEGG为8 423个。沈晔等进行脊尾白虾对低盐胁迫响应的转录组学分析,结果获得了72 734 条Unigenes,注释到NR为21 931个。由此看出,单分子实时测序技术获得的序列质量、基因数量和注释基因信息优于第二代测序。
李斌等用波纹龙虾肝胰腺和卵巢组织的mRNA表达谱进行了2代转录组测序,测序结果与该研究3代测序结果相差较大,2代测序总Unigene 74 124个,而该研究为10 425个,造成3代测序的Unigene比2代少的原因是3代测序进行了无参转录组对照测序,只能比对到其他数据库,这样测序出来的结果就远比2代的少。2代测序基因功能注释率为33.80%,该研究为93.74%;2代测序仅14.00%注释到GO数据库,22.70%注释到KEGG代谢途径,12.00%注释到COG蛋白数据库。造成这种结果的原因可能是对波纹龙虾开展研究少,国内外对波纹龙虾的研究报道也相对较少,在NCBI数据库中找到龙虾属的核酸序列不足1 000条;也有可能是个体间的差异较大和测序上样量的不同,2代测序和3代测序条件和方法有所差异。生物分子数据库的完善对波纹龙虾的研究、养殖和保护起着重要作用,因此加大波纹龙虾的分子生物学研究力度至关重要。
Zhang等通过凡纳滨对虾全长转录组文库获得72 648条高质量序列,Wang等通过对中国对虾进行全长转录组文库测序获得10 795条高质量序列。该研究通过波纹龙虾全长转录组文库最终获得10 425条高质量序列,较斑节对虾、凡纳滨对虾和中国对虾少,这可能是物种之间的差异。这些数据为进一步了解波纹龙虾的生物学特性、基因功能、相关代谢途径和信号通路等提供理论基础,为后续研究提供一定参考。
