药品中需氧菌总数能力验证不满意结果的OOS分析
2022-08-02王立云杨晓云牛萌萌姬俊
王立云 杨晓云 牛萌萌 姬俊
(青岛市食品药品检验研究院/国家药品监督管理局海洋中药质量研究与评价重点实验室 山东 青岛 266071)
1 前言
出具准确可靠的检测数据,不断提升检测能力是实验室运行的基础[1],加强内部质量控制,参与外部能力验证是评价实验室检测能力的重要手段[2],能够帮助实验室识别风险。
本实验室参与2021年度药品需氧菌总数的能力验证,在3个样品中出现1个不满意结果(样品1),2个满意结果(样品2、3),按照实验室认可质量体系的要求,对不满意结果的纠正与预防措施(Corrective Action and Protective Action,CAPA)立即全过程启动[3],本文对此进行分析。
2 处理过程
2.1 启动《不符合检验工作控制程序》
本实验室立即启动《不符合检验工作控制程序》,填写《不符合工作处理单》,终止该项目的检验工作,并及时向质量控制部门报告情况,申请进一步识别并获准停止该项工作的正式许可,分析不满意结果产生的根本原因,梳理近半年的检验报告和原始记录,提出纠正措施并实施[4-5]。
2.2 原因分析
按照《中国药典》2020版中“9203药品微生物实验室质量管理指导原则”,从人员、样品、设施环境等因素进行逐一排查,最终确定引起不满意结果的原因。
(1)人员。3名检验人员均经过上岗培训并取得资格证,有多年微生物检验工作经验。本次实验有3批样品,每人分别负责1批样品的开启、溶解,并制备待测溶液,同时进行3份样品的稀释、加培养基等操作,因3人的计数结果一致,故初步排除人员的问题。
(2)培养基。该试验采用本实验室常用的国内A厂家的TSA成品培养基,在购入后曾进行验收,结果为合格,且密闭储存于设有除湿机的房间,实验前现配,灭菌后调节pH。采用中检院的胰酪大豆胨琼脂对照培养基(用于需氧菌总数计数适用性检查或检验)作为比对培养基,结果与A厂家培养基一致,由结果报告可知,试验分到的样品中2份为同批次样品,1份结果满意,1份结果不满意,故可排除培养基的问题。
(3)试剂。该试验采用国内A厂家生产的pH7.0无菌氯化钠—蛋白胨缓冲液,每瓶500 mL,临用现开。
(4)菌种。该试验未做阳性对照,所用的TSA培养基为日常备用,曾多次验证(加标回收),均符合《中国药典》2020年版的要求,最近一次使用距离该实验未超过10 d,以空白培养基为阴性对照。
(5)设备。该试验所用培养箱为INE-500型培养箱,2021年3月1日由青岛市计量院进行检定及校正,试验前用外置温度计对数显屏幕上的温度进行核对,故可排除培养条件的问题。
(6)设施和环境条件。由结果报告可知,为考察实验室在检验过程中是否由于环境设施、操作等污染样品,故样品3中未添加菌种,本实验室检测样品3无菌,为满意结果,可以反证设施及环境条件符合要求。
(7)样品及检验方法。该试验样品为3瓶分别置于安瓿瓶中的乳白色冷冻干燥物,收到样品后第一时间置于-20℃度冰箱中保存,检测前将样品平衡至室温。制备待测溶液时,为避免转移损失,未进行转移操作,直接向安瓿瓶中加入10 mL pH7.0无菌氯化钠—蛋白胨缓冲液,充分混匀后完全溶解成乳白色均一溶液,作为待测供试品(10 mL);按作业指导书中“室温放置15 min”(为细菌能够充分复苏),再次混匀,进行10倍稀释,取待测液1 mL置于9 mL pH 7.0无菌氯化钠—蛋白胨缓冲液中;分别取待测供试液原液、1∶10液、1∶100液1 mL置于培养皿中,加入配置好的温度低于45℃的TSA培养基,混匀,静置;待培养基凝固后置于32.5℃培养箱中培养,每日点计。该试验过程较简单,偏差不应来源于此。
(8)结果计算及报告。该试验有2处易被忽视的细节:未注意作业指导书中对待测溶液的规定,误认为第一步稀释的溶液是1∶10溶液,而将上报结果扩大了10倍,即cfu/g,实际应为cfu/mL;数据上报应取菌落数小于300的稀释级别[6],计算后取2位有效数字。
由结果报告可知,样品1和2为同批样品,添加约103~104cfu的微生物。在统计本次能力验证结果时,该样品的结果中位值为1 500;在考察样品均匀性和稳定性时,该样品的含菌量约为1 000,本实验室的结果偏低,样品3中未添加微生物,结果见表1。

表1 能力验证结果
进一步分析原始数据(见表2),样品1、2由原液到1∶1 000的10倍稀释液中计数存在异常,前者含菌量明显偏低,未成梯度趋势。此时按照药典中取平均最高菌落数计算后的数值上报,结果与实际含菌量不符。在1∶100稀释级得到的结果是样品1、2中均含有1 400、1 500cfu/mL的微生物,说明样品中的含菌量达到1 000以上,原液或1∶10溶液得到的结果偏低,应是菌落未长出或未被计数。若高稀释度平板上的菌落数更高,则说明检验过程出现差错或样品中含抑菌物质,不满意结果的原因应在于此。
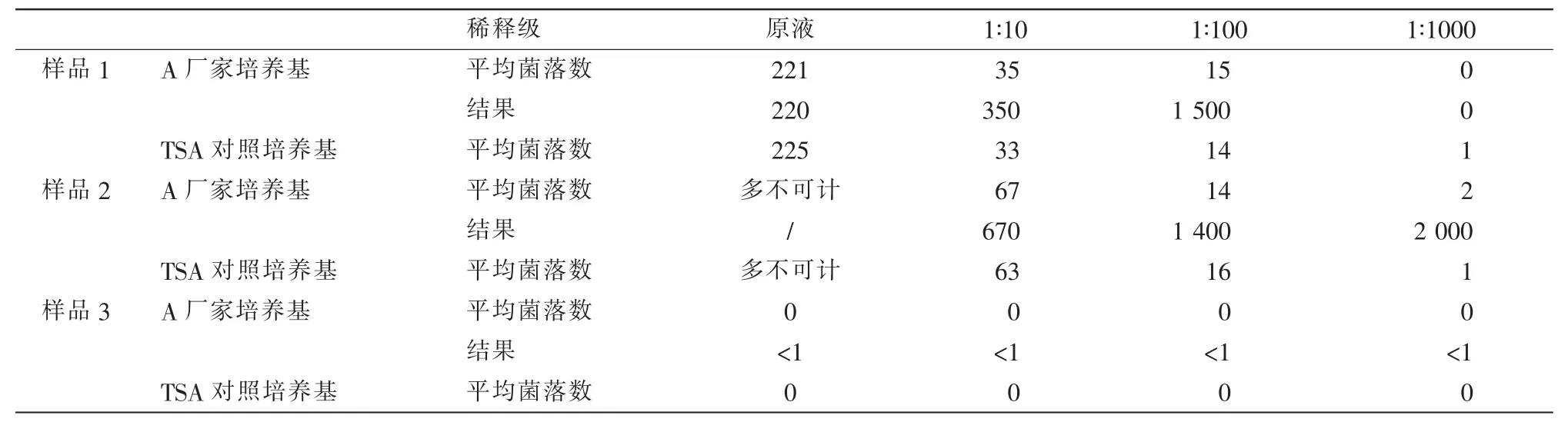
表2 能力验证原始数据
该试验样品中添加的微生物包含金黄色葡萄球菌(CMCC(B)26003)、(CMCC 63303)和白色念珠菌(CMCC(F)98001)。蜡样芽孢杆菌的菌落较大,表面粗糙、扁平、不规则,在普通琼脂平板培养基上37℃培养24 h,可形成圆形或近似圆形、质地软、无色素、稍有光泽、直径5~7 mm的白色菌落(似蜡烛样颜色)。白色念珠菌则生长较慢,部分菌落48 h才肉眼可见,若平板上的微生物多,可能会造成菌落覆盖或重叠,虽逐日点计,但易被漏数,导致结果偏低。
综上所述,可以确定本次能力验证不满意结果的原因是原液或1∶10溶液菌落未长出或未被计数,未经过全面分析,照搬中国药典的菌落计数规则上报结果造成的。
3 纠正措施
当参加能力验证活动结果为不满意或可疑时,按照本院质量管理体系要求,应立即进行检验结果偏差(Out of Specification,OOS)分析,制定纠正与预防措施(Corrective Action and Protective Action,CAPA)[7-8],并通过参加测量审核,再次进行考察直至取得满意结果。
(1)人员培训。对所有试验人员进行《中国药典》2020年版四部《1105非无菌产品微生物限度检查:微生物计数法》《中国药品检验标准操作规范》2019年版的培训,重新梳理知识体系,提高人员检验水平。
(2)检测检品。调取留样,特别是距离该试验最近一次验证的样品,重新对其进行回收试验,结果均在0.5~2.0之间,重新进行微生物限度检查,2次结果均一致,详见表3。
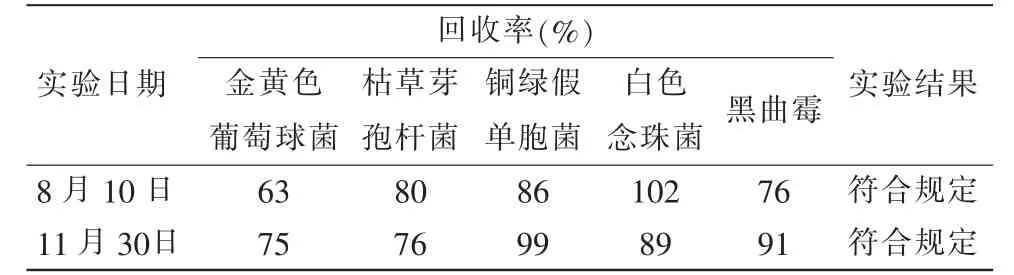
表3 能力验证前后同批次检品实验验结果对比
(3)实验室比对。从其他实验室调取样品进行比对试验,结果均一致。
(4)参加测量审核。立即报名参加药品中需氧菌总数测量审核,3批样品均得到满意结果。
4 监督、跟踪、恢复检验
纠正措施实施后,由质量监督员进行跟踪验证,确认各项纠正措施是否落实到位并按要求完成,检查实施情况是否有完整记录[8]。符合要求后,恢复检验工作,在下次的监督检查中还要进—步跟踪验证。严格审查纠正措施的可行性和有效性,对反复出现的问题加大审核的频次和力度[9]。
5 讨论
本次能力验证的组织方共制备了3种浓度6批样品,分别为能力验证样品A1、A2,B1、B2,C1、C2,合计制备样品总数为955瓶。样品A1、A2添加约105cfu的微生物,样品B1、B2添加约103~104cfu的微生物,样品C1、C2不添加微生物。组织方向每个实验室发放3份样品,为保证考核难度的均衡性,均包含1份样品C1或C2,剩余2份样品从样品A1、A2、B1、B2中随机抽取。
本次能力验证中,170家实验室的结果为满意,占比76.2%;10家实验室结果为可疑,占比4.5%;43家实验室结果为不满意,占比19.3%。经统计,A、B这2类样品中,有62份样品得到可疑或不满意的结果,其中B2样品的可疑或不满意率占比33.87%,C类样品未计入,结果见表4。可见B类样品的可疑或不满率大于A类样品。这可能与样品中微生物的数量有关,8月中下旬我国大部分地区高温高湿,微生物易受运输、温度等条件的影响。

表4 样品A1、A2、B1、B2(可疑+不满意)占比
能力验证是实验室质量控制的重要手段之一,是评价实验室技术能力的重要依据,实验室通过有目的、有计划地参加能力验证,可提高检验人员的检验能力。任何一个步骤出现问题都可能导致最终结果的偏离,因此验证结束后,组织方会提供详细的《能力验证结果报告》,对本次计划的关键技术要点进行详尽分析,能够促进检验水平的提升[10]。能力验证的结果若出现可疑或不满意,应当通过纠正与预防措施(CAPA)查找原因,并进行整改,以提升实验室的检验能力和管理水平。
