扇贝组织中不同形态氨基脲测定方法研究
2022-08-01邢丽红孙伟红朱盼盼孙晓杰李兆新
邢丽红, 孙伟红, 朱盼盼, 孙晓杰, 李兆新
扇贝组织中不同形态氨基脲测定方法研究
邢丽红1, 2, 3, 孙伟红1, 2, 3, 朱盼盼1, 2, 3, 孙晓杰1, 2, 3, 李兆新1, 2, 3
(1. 中国水产科学研究院 黄海水产研究所 农业农村部水产品质量安全检测与评价重点实验室, 山东 青岛 266071; 2. 青岛海洋科学与技术试点国家实验室, 山东 青岛 266033; 3. 大连工业大学 海洋食品精深加工关键技术省部共建协同创新中心, 辽宁 大连 116034)
为明确环境新型污染物--氨基脲(semicarbazide, SEM)在贝类体内的存在形态, 本文建立了一种SEM在扇贝中形态测定的液相色谱-串联质谱(liquid chromatography-tandem mass spectrometry, LC-MS/MS)分析方法。扇贝中SEM总量测定方法为: 样品用盐酸水解, 以2-硝基苯甲醛为衍生剂衍生, 乙酸乙酯提取, 超滤离心净化, 进LC-MS/MS分析。结合态SEM测定方法为: 样品分别采用50%甲醇水溶液、75%甲醇水溶液、甲醇和水洗涤后, 按照SEM总量测定方法处理, 采用甲醇和2 mmol/L乙酸铵作为流动相进行梯度洗脱, 电喷雾离子源正离子(electrosprary ionization, ESI+)选择反应监测模式对SEM进行定性和定量测定。本方法在1~20 μg/L范围内线性关系良好, 相关系数大于0.999。SEM在1.00、5.00、10.0 μg/kg添加水平的回收率在84.2%~118%, 相对标准偏差均<15%。本方法扇贝中SEM的定量限为1.0 μg/kg, 本方法灵敏、准确、操作简便, 适用于扇贝中SEM存在形态的测定。
氨基脲; 液相色谱-串联质谱法; 扇贝; 存在形态
氨基脲(semicarbazide, SEM)是呋喃西林药物在动物体内的特征代谢产物, 由于呋喃西林原型药物在动物体内代谢快速, 因此通常以检测SEM来判定是否非法使用呋喃西林[1-3]。欧盟、美国等发达国家和中国先后制定了禁止使用该类药物的规定[4-6]。欧盟2003/181/EC规定硝基呋喃类代谢物在各种检测方法中的灵敏度为1 μg/kg[7]。中国在水产品质量安全监测中以1 μg/kg作为硝基呋喃类代谢物的判定限量。但近年来研究表明, 呋喃西林的代谢降解并非是水产品中SEM残留的唯一来源, 很多途径均可以引入SEM污染[8]。偶氮二甲酰胺作为一种塑料发泡剂, 常用在与食品接触的材料中, 其在高温下可分解产生SEM[9-10]; 偶氮二甲酰胺作为面粉发泡剂, 在高温条件下可分解产生SEM[11-13]; 食品经次氯酸盐处理可产生SEM[14]; SEM天然存在于一些动物和植物体内[2, 15-16]。各种途径产生的SEM均可能排入环境, 成为环境中的一种污染物, 污染水生生物赖以生存的环境, 最终在其体内蓄积。目前有文献报道部分海域的海水、海底沉积物已经受到不同程度的污染, 同时在海洋生物体内检测到SEM残留[17-20]。贝类通过富集海水中的SEM, 从而影响该类产品的质量安全, 并通过食物链对人体健康产生危害。
呋喃西林进入动物体内后能够快速代谢, 其代谢产物SEM主要以结合态形式在生物体内长期稳定存在[21-23], 结合态SEM通过常用的食品烹饪方法如蒸煮、煎炸、烘烤和微波加热等方式均无法使其完全降解[24], 而游离态SEM则可通过洗涤等方式去除[25-26]。贝类产品对SEM具有一定的富集能力, 然而环境中的SEM进入贝类体内究竟是以何种形态存在尚不明确。SEM常用的测定方法有高效液相色谱法[27-28]、液相色谱-串联质谱法[29-30]、酶联免疫分析[31-32]、胶体金免疫分析法[33]等。现有检测方法尚无针对贝类产品中SEM形态的测定方法。本文通过优化样品前处理方法, 建立了贝类组织中SEM存在形态的液相色谱-串联质谱测定方法, 本研究为分析环境污染物SEM在贝类产品中的存在形态奠定良好的基础, 同时也为评估贝类产品中氨基脲含量提供了技术支撑。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器与设备
AB 5500 Qtrap LC-MS/MS仪(配岛津LC20 HPLC仪, 美国AB SCIEX公司); 分析天平(感量0.01 g, 德国赛多利斯集团); Vortex 2涡旋混合器(德国IKA公司); IS-RDS3恒温振荡器(美国精骐有限公司); Himac GR22G高速离心机(日本日立公司); N-EVAP 112氮气吹干仪(美国Organomation 公司); 微量高速离心机(德国Sigma公司); Gradient A10 Mill-Q超纯水仪(美国Millipore公司)。
1.1.2 材料与试剂
SEMHCl标准品(纯度99.8%, Dr.Ehrenstorfer GmbH公司); SEM-13C,15N2(纯度99.2%, WiTEGA公司); 甲醇(色谱纯, Merck公司); 盐酸(优级纯, 国药集团化学试剂有限公司); 二甲亚砜(色谱纯, CNW公司); 2-硝基苯甲醛(纯度≥98%, CNW公司); 磷酸氢二钾(优级纯, 国药集团化学试剂有限公司); 乙酸乙酯(色谱纯, Merck公司); 乙酸铵(色谱纯, Merck公司); 1.5 mL超滤离心管(10 K, Millopore)。
1.2 试验方法
1.2.1 溶液配制
SEM标准储备液: 准确称取适量SEMHCl标准品, 用甲醇溶解并定容至10 mL棕色容量瓶中, 配制成1.0 mg/mLSEM标准储备液, –20 ℃避光保存。SEM-13C,15N2内标标准储备液: 准确称取适量SEM-13C,15N2标准品, 用甲醇溶解并定容至10 mL棕色容量瓶中, 配制成1.0 mg/mL内标标准储备液, –20 ℃避光保存。使用时分别量取适量SEM、SEM-13C,15N2标准储备液置于棕色容量瓶中, 用甲醇逐级稀释, 分别配制成1 μg/mL的SEM标准工作液和100 ng/mL内标标准工作液。
1.2.2 阳性基质的制备
将扇贝暴露在含有SEM的海水中进行养殖并采集扇贝样品, 将样品外壳清洗后开壳剥离, 分别取闭壳肌、内脏、外套膜、性腺和鳃组织, 匀浆后于–20 ℃避光保存, 用于结合态SEM测定方法研究。
1.2.3 SEM总量测定
1.2.3.1 水解和衍生化
称取扇贝闭壳肌、内脏、外套膜、性腺和鳃组织2 g, 置于50 mL离心管中, 准确加入内标标准工作液50 μL, 充分涡旋混合1 min, 再加入0.5 mol/L盐酸溶液5 mL和50 mmol/L 2-硝基苯甲醛溶液150 μL, 充分涡旋混合1 min后, 置于恒温振荡器中, 在37 ℃条件下避光振荡16 h。
1.2.3.2 提取和净化
将离心管取出并冷却恢复至室温后, 加入1.0 mol/L磷酸氢二钾溶液, 调节pH至7.0~7.5。加入8 mL乙酸乙酯, 充分涡旋振荡1 min, 以8 000 r/min高速离心5 min。转移上层清液至10 mL离心管中, 于40 ℃下氮气吹干。准确加入5%甲醇溶液1.0 mL, 充分涡旋振荡溶解残留物, 再将溶液转移至超滤离心管中, 14 000 r/min离心10 min, 取下层清液进LC-MS/MS分析。
1.2.4 结合态SEM测定
称取扇贝组织2 g于50 mL离心管中, 加入50%甲醇水溶液5 mL, 充分涡旋混合50 s, 超声波提取5 min, 再以4 000 r/min离心10 min, 弃去上清液。继续分别依次加入5 mL 75%甲醇水溶液、5 mL甲醇和5 mL超纯水, 重复上述操作, 弃去上清液, 再按照上述步骤1.2.3处理样品。
1.2.5 液相色谱-串联质谱条件
1.2.5.1 液相色谱条件
Waters Xbridge C18色谱柱(150 mm×2.1 mm, 3.5 μm); 流速: 0.35 mL/min; 流动相: A. 2 mmol/L乙酸铵溶液, B. 甲醇, 梯度洗脱程序: 0~0.5 min, 90% A; 0.5~4.0 min, 90%~5% A; 4.0~5.5 min, 5% A; 5.5~6.0 min, 5%~90% A; 6.0~7.0 min, 90% A; 进样量: 10 µL; 柱温: 35 ℃。
1.2.5.2 质谱条件
离子源: 电喷雾离子源, 正离子模式; 喷雾电压: 5 500 V; 气帘气: 30 psi; 雾化器: 35 psi; 辅助加热气: 35 psi; 离子源温度: 550 ℃; 碰撞气: Medium; 扫描模式: 多反应选择监测; 选择反应监测母离子、子离子和碰撞能量等质谱参数见表1。
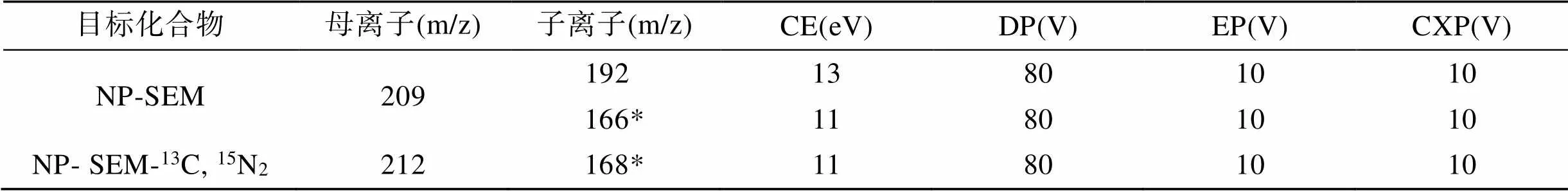
表1 SEM及其内标的质谱检测参数表
注: *为定量碎片离子
2 结果与讨论
2.1 结合态SEM测定方法研究
样品中结合态SEM的测定方法为: 先将游离态SEM通过洗涤方式去除, 再将结合态SEM水解衍生化测定。本研究分别以不同极性的洗涤溶剂处理贝类阳性基质样品的不同组织, 研究洗涤方式对测定结果的影响。其中, A处理组为未采用洗涤方式处理样品, 直接测定SEM总量; B处理组为采用50%甲醇水溶液洗涤1次样品; C处理组为采用50%甲醇水溶液和75%甲醇水溶液各洗涤1次样品; D处理组为分别采用50%甲醇水溶液、75%甲醇水溶液和甲醇溶液各洗涤1次样品; E处理组为分别采用50%甲醇水溶液、75%甲醇水溶液、甲醇溶液和水溶液分别各洗涤1次样品。B、C、D、E处理组分别采用不同极性试剂处理样品, 其SEM测定结果见图1。由图1可知, 采用B方法处理扇贝组织样品时, 洗涤1次即可去除闭壳肌、性腺、内脏、外套膜和鳃中65.5%、69.3%、34.2%、65.6%、53.7%游离态SEM。继续增加洗涤试剂和洗涤次数, 即可洗涤去除样品组织中剩余的游离态SEM, 但SEM含量下降趋势减缓。不同处理组间SEM差异比较采用SPSS18.0软件进行统计分析, 差异性分析采用Mann-Whitney U检验。通过组间数据显著性差异分析结果表明, A、B、C、D 4个处理组间均存在显著性差异(<0.05), 而D处理组和E处理组间无显著性差异(>0.05)。由以上分析可知, 通过改变洗涤溶剂的极性, 可以去除样品组织中游离态SEM, 游离态SEM在各组织中所占比例较高。当采用E处理方法洗涤样品时, SEM测定结果与D处理组无显著性差异。因此本实验采用E处理方法分析扇贝不同组织中结合态SEM。
2.2 基质效应对SEM测定结果的影响
基于液质联用法分析生物样本的目标化合物时, 样本的一些共同提取物可能对目标化合物的离子化效率产生增强或者抑制效应, 即基质效应。基质效应是影响药物残留检测分析结果准确度的重要因素, 本试验分别对SEM总量和结合态SEM的基质效应进行了考察。

图1 不同洗涤方法对SEM测定结果的影响(n=5)
a、b、c、d. 在0.05水平上有显著性差异; A. SEM总量; B.50%甲醇水; C. 50%甲醇水+75%甲醇水; D. 50%甲醇水+75%甲醇水+甲醇; E. 50%甲醇水+75%甲醇水+甲醇+H2O
a, b, c, d.significant difference (< 0.05); A. Total SEM; B. 50% methanol-water solution; C. 50% methanol-water solution, and 75% methanol-water solution; D. 50% methanol-water solution, 75% methanol-water solution, and methanol solution; E. 50% methanol- water solution, 75% methanol-water solution, methanol solution, and water
2.2.1 基质效应对SEM总量测定结果的影响
按照1.2.3的方法处理扇贝组织, 分析基质效应对SEM总量测定结果的影响。基质效应(ME)可通过同浓度提取液中SEM峰面积响应值(B)与纯溶剂中SEM峰面积响应值(A)的比值来考察, 即ME=B/A。当ME>1时, 表示基质对SEM的响应具有增强效应; 当ME=1时, 说明不存在基质效应; 当ME<1时, 则表示基质对SEM的响应具有抑制效应。SEM及其内标在纯溶液中的响应值见表2, SEM在基质提取液中的响应值见表3。
由表2和表3可知, 在5种基质标准溶液中, 同浓度SEM的峰面积均小于标准溶液中SEM的峰面积, ME值在0.419~0.751, 说明基质对SEM响应具有抑制效应。而在同浓度下, 基质溶液中SEM与相应内标峰面积的比值和标准溶液中SEM与内标峰面积的比值基本一致。以SEM与SEM-13C,15N2峰面积的比值为纵坐标, 以SEM浓度为横坐标进行线性拟合, 拟合后的校正曲线方程见表4。SR表示基质匹配校正曲线方程斜率与溶剂标准溶液校正曲线方程斜率的比值。由表4可知, SR值在1.018~1.163, 说明即使采用内标法也不能完全消除基质效应的影响, 不同基质对SEM总量测定结果具有一定程度的影响。因此, 采用基质校正曲线定量, 测定结果更加精准。

表2 SEM及其内标在标准溶液中的响应

表3 基质效应对SEM总量测定结果的影响
2.2.2 基质效应对结合态SEM测定结果的影响
按照1.2.4的方法处理扇贝组织, 通过基质提取液中SEM峰面积与纯溶剂中SEM峰面积的比值来考察基质效应(ME)。
采用1.2.4方法通过洗涤处理将扇贝组织中的游离态SEM和杂质成分一同去除, 与SEM总量测定方法相比, 其基质效应存在差异。由表5可知, 在同一浓度下, 外套膜和鳃基质提取液中SEM的响应值大于标准溶液中SEM的响应值, 因此基质对SEM响应具有增强效应; 而闭壳肌、性腺和内脏基质提取液中SEM的响应值小于标准溶液中SEM的响应值, 因此具有基质抑制效应。由表6可知, SR在0.957~1.035, 说明测定结合态SEM时, 内标法基本消除基质效应的影响, 采用溶剂校正曲线, 即可准确定量测定结果。

表5 基质效应对结合态SEM测定结果的影响

表6 结合态SEM测定法校正曲线
杨婷婷等[34]对蛋及蛋制品中硝基呋喃类代谢物的残留量进行分析时, 发现不同样品基质表现出明显的基质抑制效应, 因此在实际检测时建议采用基质加标曲线定量。本研究通过对SEM总量和结合态SEM的基质效应进行考察, 结果表明, 不同形态SEM采用不同方式定量, 可使结果更加精准。
2.3 质谱条件的选择
SEM的相对分子质量为75, 质量数较小, 在该质荷比范围内, 质谱有较大的背景干扰, 且直接测定离子化效率低下, 没有特征离子碎片, 效果并不理想。通过衍生化的方法, 可以增加分子质量, 降低背景干扰。2-硝基苯甲醛是最常用的衍生化试剂, 2-硝基苯甲醛与SEM的自由氨基团衍生化生成硝基苯衍生物, 可增加分子质量, 增加离子碎片的选择性, 从而提高质谱响应。将SEM和SEM-13C,15N2标准溶液按照1.2.3的方法进行衍生化和提取, 得到NP-SEM、NP-SEM-13C,15N2标准溶液。SEM的分子中含有氨基, 属于强极性物质, 容易形成稳定的[M+H]+分子离子, 因此在正离子模式下具有较高的灵敏度。将衍生化后的标准溶液在正离子模式下进行母离子全扫描, 确定分子离子峰。以分子离子峰为母离子进行子离子扫描, 并优化得到目标化合物母离子和子离子所需的碰撞能量、去簇电压、射入电压、碰撞室射出电压等质谱参数。NP-SEM的两个子离子中,/196是丰度最大的子离子, 但在其出峰处存在杂质干扰, 因此选择次强碎片离子/166作为定量离子,/196作为定性离子。本方法中NP-SEM、NP-SEM-13C,15N2的母离子、定性离子和定量离子及碰撞能量等质谱参数见表1。
2.4 净化条件的选择
扇贝组织样品基质较为复杂, 在用乙酸乙酯进行液液萃取的操作步骤中, 当以4 000 r/min离心时, 萃取液可能会由于蛋白、脂类等干扰而形成乳化层。将该溶液重新涡旋混匀后, 提高转速, 以8 000 r/min离心, 可有效避免乳化层的形成, 并有利于萃取液的转移。在40 ℃下氮气浓缩至干后, 加入5%甲醇水溶液溶解残渣, 由于基质的作用, 可能会出现浑浊。将待测液转移至超滤离心管中, 经14 000 r/min超滤离心净化作用, 杂质干扰物质与待测液可实现较好的分离状态, 取下层清液进液相色谱-串联质谱仪分析, 操作既简便快捷, 又可以避免杂质干扰, 取得较好的实验结果。
现有硝基呋喃类代谢物的净化方法主要有固相萃取净化[35]、基质分散固相萃取净化[36]等方式。固相萃取净化方法操作步骤繁琐, 批量操作费时费力。而基质分散固相萃取净化方法对基质成分复杂的样品, 净化效果不太理想。本方法采用超滤净化, 极大简化了操作步骤, 又可取得很好的净化效果。
2.5 方法的灵敏度
采用在空白样品中添加标准溶液的方法可确定本方法的灵敏度。将SEM标准溶液添加到空白贝类组织中, 按照样品水解、衍生化、提取和净化等步骤处理样品, 采用1.2.5的色谱和质谱条件进LC-MS/ MS分析。当添加水平分别为0.5和1.0 μg/kg时, SEM信噪比分别≥3和≥10, 由此确定本方法SEM的检出限为0.5 μg/kg, 定量限为1.0 μg/kg。扇贝组织中SEM及其同位素内标特征离子质量色谱图见图2。
2.6 方法的准确度及精密度
方法的准确度和精密度分别采用回收率和相对标准偏差表示。扇贝组织中SEM总量包括游离态SEM和结合态SEM, 结合态SEM的测定方法是先用不同极性的溶剂将组织中的游离态SEM洗涤去除, 剩余SEM即为蛋白结合态SEM, 然后再按照与SEM总量相同的测定步骤进行分析。因此本方法的添加回收率试验是在空白扇贝组织中添加SEM标准溶液, 然后按照SEM总量的测定方法进行添加回收率分析的。在空白扇贝组织中, 按照低、中、高浓度添加SEM, 添加水平分别为1.00、5.00、10.0 μg/kg, 每个添加水平平行测定5次, 计算每种组织的回收率和精密度, 其测定结果见表7。
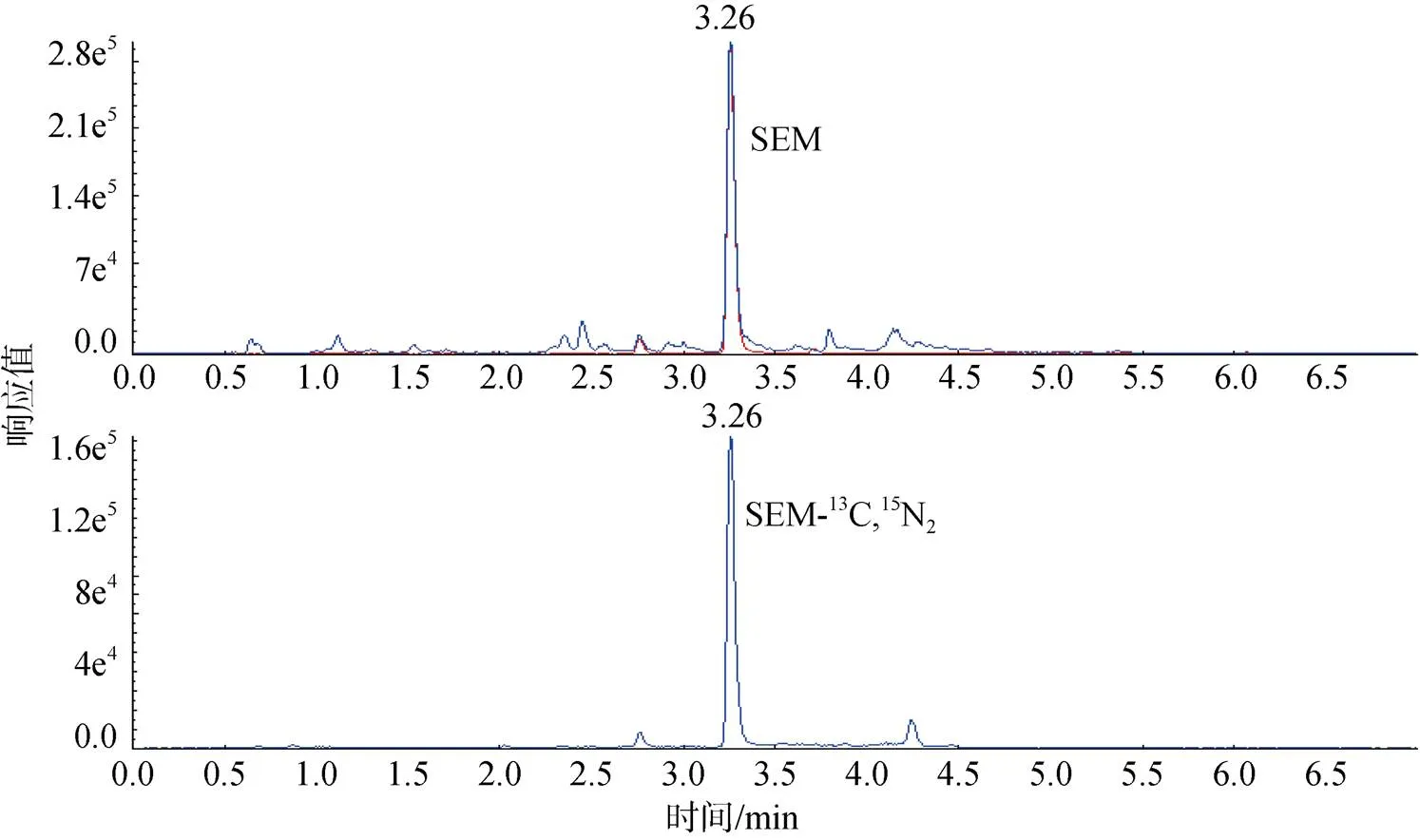
图2 扇贝闭壳肌中SEM及其同位素内标的特征离子质量色谱图
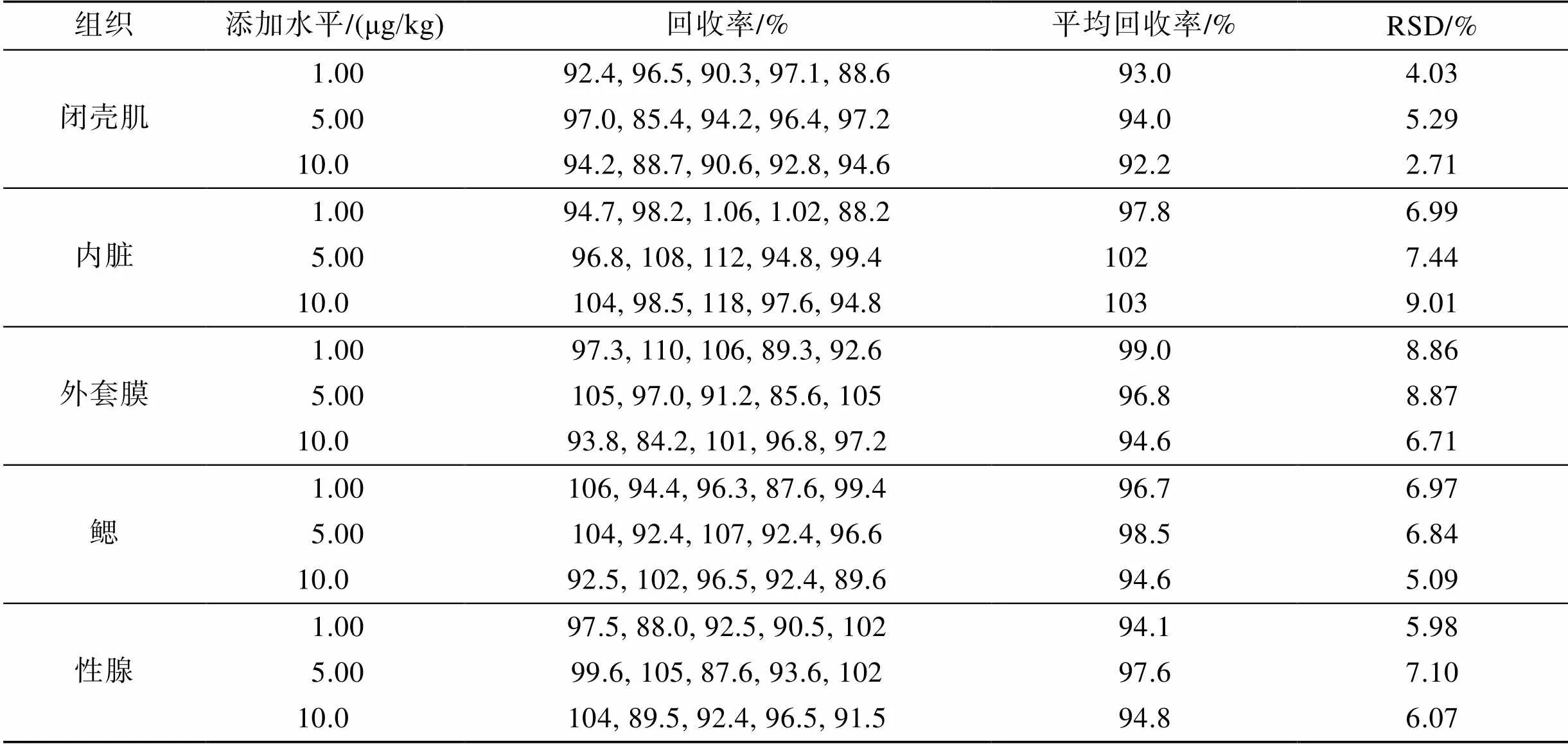
表7 扇贝组织中添加SEM的回收率(n=5)
在1.00、5.00和10.0 μg/kg 3个添加水平下, 测得SEM的回收率在84.2%~118%, 由此说明, 该方法具有良好的准确度。贝类不同组织在3个添加水平下分别平行测定5次, 测定结果的相对标准偏差均<15%, 说明本方法精密度良好。
2.7 实际样品形态测定
采用1.2.3和1.2.4建立的方法分别对本实验制备的阳性扇贝组织中SEM总量和结合态SEM含量进行测定, 结果表明, 闭壳肌、内脏、外套膜、鳃和性腺中结合态SEM分别占12.6%、32.5%、18.5%、29.6%、16.6%, 采用该方法可以对扇贝组织中SEM总量和结合态SEM含量进行准确定性定量测定。
关于SEM在水产品中存在形态的研究报道目前还较少。VAN等[26]研究了SEM在虾中的存在形态,罗氏沼虾()在含50 mg/L呋喃西林的养殖水体中暴露1周, 游离态SEM和结合态SEM均有显著增加, 肌肉组织中结合态SEM约是SEM总量的1/4, 而壳中结合态SEM比例超过65%。舒秀君等[37]研究发现, 日本沼虾()肌肉和肝胰腺中内源性SEM主要以游离态存在, 而甲壳、眼柄、附肢、头胸部和鳃组织中内源性SEM则主要以结合态存在。SEM作为一种海洋环境污染物, 其在贝类体内的存在形式尚不明确, 本文建立了扇贝体内SEM存在形态的测定方法, 为进一步研究环境污染物SEM在贝类体内的存在形态提供了技术支撑。
3 结论
本文研究了扇贝组织中SEM存在形态的测定方法, 考察了基质效应对SEM总量和结合态SEM测定结果的影响, 优化了样品净化方法, 建立了扇贝中SEM存在形态的测定方法, 该方法具有良好的准确度和精密度, 为研究SEM在贝类产品中存在形态提供了简便、灵敏、准备可靠的技术方法。
[1] JUSTIN G B. Semicarbazide is non-specific as a marker metabolite to reveal nitrofurazone abuse as it can form under Hofmann conditions[J]. Food Addit Contam, 2009, 26(1): 47-56.
[2] 于慧娟, 李冰, 蔡友琼, 等. 液相色谱-串联质谱法测定甲壳类水产品中氨基脲的含量[J]. 分析化学, 2012, 40(10): 1530-1535.
YU Huijuan, LI Bing, CAI Youqiong, et al. Determination of semicarbazide content in crustaceans by liquid chromatography tandem mass spectrometry[J]. Chin J Anal Chem, 2020, 40(10): 1530-1535.
[3] STADLER R H, VERZEGNASSI L, SEEFELDER W, et al. Why semicarbazide (SEM) is not an appropriate marker for the usage of nitrofurazone on agricultural animals[J]. Food Addit Contam A, 2015, 32(11): 1842-1850.
[4] Commission Regulation (EC) No 1442/95. Amending annexesⅠ, Ⅱ, Ⅲand IV of Council Regulation (EEC) No 2377/90 laying down a community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin[S]. Official J European Communities, 1995, 143: 26-30.
[5] FDA. Topical nitrofurans; extralabel animal drug use; order of prohibition[J]. Fed Regist Dep Heal Hum Serv, 2002, 67: 5470-5471.
[6] 畜牧兽医局. 农业农村部公告第235号动物性食品中兽药最高残留量限量[S]. 中华人民共和国农业农村部, 2002.
Animal Husbandry and Veterinary Bureau. Announcement No.235 of the Ministry of Agriculture of China. Maximum residue limits of veterinary drugs in animal food[S]. The Ministry of Agriculture of the People’s Republic of China, 2002.
[7] Commission Decision (2003/181/EC). Amending decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin[S]. Official J European Union, 2003, 71: 17-18.
[8] YU W L, LIU W H, TIAN W R, et al. Semicarbazide universality study and its speculated formation pathway[J]. J Food Safety, 2019, 39(1): 1-8.
[9] DE SOUZA S V C, JUNQUEIRA R G, GINN R. Analysis of semicarbazide in baby food by liquid chromatography tandem mass spectrometry (LC-MS-MS): in house method validation[J]. J Chromatogr A, 2005, 1077(2): 151-158.
[10] VASS M, DIBLIKOVA I, CERNOCH I, et al. ELISA for semicarbazide and its application for screening in food contamination[J]. Anal Chim Acta, 2008, 608(1): 86-94.
[11] ADAM B, BENJAMIN P Y L, DAVID L, et al. Semicarbazide formation in azodicarbonamide-treated flour: A model study[J]. J Agrci Food Chem, 2004, 52: 5730- 5734.
[12] YE J, WANG X H, SANG Y X, et al. Assessment of the determination of azodicarbonamide and its decomposition product semicarbazide: investigation of variation in flour and flour products[J]. J Agric Food Chem, 2011, 59(17): 9313-9318.
[13] CHEN L, CUI H, DONG Y L, et al. Simultaneous detection of azodicarbonamide and the metabolic product semicarbazide in flour by capillary electrophoresis[J]. Food Anal Method, 2016, 9(5): 1106-1111.
[14] HOENICKE K, CATERMANN R, HARTIG L, et alFormation of semicarbazide (SEM) in food by hypochlorite treatment: is SEM a specific marker for nitrofurazone abuse?[J]. Food Addit Contam, 2004, 21(6): 526-537.
[15] MCCRACKEN R, HANNA B, ENNIS D, et al. The occurrence of semicarbazide in the meat and shell of Bangladeshi fresh-water shrimp[J]. Food Chem, 2013, 136(3/4): 1562-1567.
[16] KWON J W. Semicarbazide: Natural occurrence and uncertain evidence of its formation from food processing[J]. Food Control, 2017, 72: 268-275.
[17] XU Y J, SUN X K, TIAN X H, et al. Survey of semicarbazide contamination in coastal waters adjacent to the chaohe river estuary[J]. Oceanologia Et Limnologia Sinica, 2010, 41(4): 1-5.
[18] 于召强, 徐英江, 田秀慧, 等. 四十里湾海洋贝类对氨基脲的生物富集特性[J]. 海洋环境科学, 2013, 32(1): 39-42.
YU Zhaoqiang, XU Yingjiang, TIAN Xiuhui, et al. Semicarbazide bioaccumulation in seashells of Sishili Bay[J]. Marine Envieonment Science, 2013, 32(1): 39-42.
[19] TIAN X H, XU Y J, SONG X K, et al. Temporal and spatial distribution of semicarbazide in western Laizhou Bay[J]. Mar Pollut Bull, 2016, 112: 393-398.
[20] TIAN X H, XU Y J, GONG X H, et al. Environmental status and early warning value of the pollutant semicarbazide in Jincheng and Sishili Bays, Shandong Peninsula, China[J]. Sci Total Environ, 2017, 576: 868-878.
[21] NOUWS J F, LAURENSEN J. Postmortal degradation of furazolidone and furaltadone in edible tissues of calves[J]. Vet Q, 1990, 12: 56-59.
[22] KIM D, KIM B, HYUNG S W, et al. An optimized method for the accurate determination of nitrofurans in chicken meat using isotope dilution–liquid chromatography/mass spectrometry[J]. J Food Compos Anal, 2015, 40: 24-31.
[23] YUAN G X, ZHU Z, YANG P, et al. Simultaneous determination of eight nitrofuran residues in shellfish and fish using ultra-high performance liquid chromatography–tandem mass spectrometry[J]. J Food Compos Anal, 2020, 92: 103540.
[24] COOPER K M, KENNEDY D G. Stability studies of the metabolites of nitrofuran antibiotics during storage and cooking[J]. Food Addit Contam, 2007, 24: 935-942.
[25] CHU P S, LOPEZ M I. Liquid chromatography-tandem mass spectrometry for the determination of protein- bound residues in shrimp dosed with nitrofurans[J]. J Agric Food Chem, 2005, 53: 8934-8939.
[26] VAN POUCKE C, DETAVERNIER C, WILLE M, et al. Investigation into the possible natural occurence of semicarbazide inprawns[J]. J Agric Food Chem, 2011, 59: 2107-2112.
[27] WANG K, KOU Y, WANG M, et al. Determination of nitrofuran metabolites in fish by ultraperformance liquid chromatography-photodiode array detection with thermostatic ultrasound-assisted derivatization[J]. ACS Omega, 2020, 5: 18887-18893.
[28] DU N N, CHEN M M, SHEN L Q, et al. Determination of nitrofuran metabolites in shrimp by high performance liquid chromatography with fluorescence detection and liquid chromatography–tandem mass spectrometry using a new derivatization reagent[J]. Journal of Chromatography A, 2014, 1327: 90-96.
[29] EUNCHAE R, JI-SUNG P, SIB SANKAR G, et al. A simplified modification to rapidly determine the residues of nitrofurans and their metabolites in aquatic animals by HPLC triple quadrupole mass spectrometry[J]. Environmental Science and Pollution Research International, 2020, 28(6): 7551-7563.
[30] 邢丽红, 孙伟红, 彭吉星, 等. 液相色谱-串联质谱法测定贝类组织中硝基呋喃类代谢物残留量[J]. 环境化学, 2019, 38(2): 287-296.
XING Lihong, SUN Weihong, PENG Jixing, et al. Determination of nitrofuran metabolites residues in shellfish tissues by high performance liquid chromatography-tandem mass spectrometry[J]. Environmental Chemistry, 2019, 38(2): 287-296.
[31] WU W, YANG S, LIU J, et al. Progress in immunoassays for nitrofurans detection[J]. Food Agric Immunol, 2020, 31: 907-926.
[32] 区兑鹏, 卢义博, 严忠雍, 等. 酶联免疫法快速检测水产品中硝基呋喃类代谢物、氯霉素及氟苯尼考[J]. 中国渔业质量与标准, 2021, 11(1): 27-33.
QU Duipeng, LU Yibo, YAN Zhongyong, et al. Enzyme-linked immunoassay for the rapid detection of furan, chloramphenicol and florfenicol in aquatic products[J]. Chinese Fishery Quality and Standards, 2021, 11(1): 27-33.
[33] 中华人民共和国国家市场监督管理总局. SN/T 4541.1- 2016 商品化试剂盒检测方法硝基呋喃类方法一[S]. 北京: 中国标准出版社, 2016.
General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China. SN/T 4541.1-2016 Commercial kit method – Nitrofurans-Test method I[S]. Beijing: Standards Press of China, 2016.
[34] 杨婷婷, 易路遥, 熊雯, 等. QuEChERS-四极杆飞行时间串联质谱法测定7种蛋及蛋制品中硝基呋喃类代谢物残留量的基质效应[J]. 肉类工业, 2018, 4: 45- 50.
YANG Tingting, YI Luyao, XIONG Wen, et al. Determination of matrix effect of nitrofuran metabolites residues in seven kinds of eggs and egg products by QuEChERS-quadrupole time-of-flight series mass spectrometry[J]. Meat Industry, 2018, 4: 45-50.
[35] 陈剑刚, 白艳玲, 梁素丹, 等. 固相萃取-液相色谱-串联质谱法测定水产品中硝基呋喃类代谢物[J]. 中国食品卫生杂志, 2013, 25(4): 52-57.
CHEN Jiangang, BAI Yanling, LIANG Sudan, et al. Determination of nitrofuran metabolites in aquatic products by high performance liquid chromatography tandem mass spectrometry with solid phase extraction[J]. Chinese Journal of Food Hygiene, 2013, 25(4): 52-57.
[36] 赵浩军, 王勇杰, 陈朋云, 等. 基质分散固相萃取与液相色谱-串联质谱联用法检测淡水鱼中硝基呋喃类代谢物的残留量[J]. 食品安全质量检测学报, 2020, 11(3): 98-103.
ZHAO Haojun, WANG Yongjie, CHEN Pengyun, et al. Detection of nitrofuran metabolites in freshwater fish by matrix-dispersed solid-phase extraction coupled with liquid chromatography-tandem mass spectrometry[J]. Journal of Food Safety & Quality, 2020, 11(3): 98-103.
[37] 舒秀君, 程波, 徐娟娟, 等. 日本沼虾养殖过程中氨基脲存在特征研究[J]. 淡水渔业, 2020, 50(3): 11-16.
SHU Xiujun, CHENG Bo, XU Juanjuan, et al. Study on the characteristics of semicarbazide (SEM) induring the culture period[J]. Freshwater Fisheries, 2020, 50(3): 11-16.
Determination method of the semicarbazide existing form in scallop
XING Li-hong1, 2, 3, SUN Wei-hong1, 2, 3, ZHU Pan-pan1, 2, 3, SUN Xiao-jie1, 2, 3, LI Zhao-xin1, 2, 3
(1. Key Laboratory of Testing and Evaluation for Aquatic Product Safety and Quality, Ministry of Agriculture and Rural Affairs, China; Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China; 2. Pilot National Laboratory for Marine Science and Technology, Qingdao 266003, China; 3. Collaborative Innovation Center of Seafood Deep Processing, Dalian Polytechnic University, Dalian 116034, China)
In order to clarify the existing form of a new environmental pollutant semicarbazide (SEM) in shellfish, a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method was developed to determine the existing form of the environmental pollutant semicarbazide (SEM) in scallop. The total SEM in scallop was determined as follows: After hydrolysis by hydrochloric acid, the sample was derivatized by 2-nitrobenzaldehyde overnight for 16 hours. Then, SEM was extracted with ethyl acetate, purified by ultrafiltration, and analyzed using LC–MS/MS. The tissue-bound SEM was separately washed with methanol/water (50: 50; v/v), followed by methanol/water (75: 25; v/v), methanol, and water. Then, the sample was determined as the method of the total SEM. The SEM residue in the extract was separated on a reversed phase using a gradient elution program of methanol and 2 mmol/L ammonium acetate. Using LC–MS/MS (electrospray ionization, ESI+) with selected reaction monitoring, identification of the major components of the SEM residue was performed based on the fragment intensities. The calibration curve showed good linearity from 0.5–20 μg/L, with a correlation coefficient over 0.999. The recoveries ranged from 84.2% to 118% for the SEM residues, with 3 spiked levels at 1.00, 5.00, and 10.0 μg/kg. The relative standard deviations were less than 15%, and the limit of quantitation for the SEM in scallop tissue was 1.0 μg/kg. The proposed method is sensitive, accurate, and easy to operate, which is suitable for determining the existing form of SEM in scallop.
SEM; high-performance liquid chromatography–tandem mass spectrometry; scallop; existing form
Nov. 19, 2021
[Supported by the National Natural Science Foundation of China, No. 41806148; Joint Development Fund Project of Zhejiang Key Lab of Exploitation and Preservation of Coastal Bio-Resource and Wenzhou Key Laboratory of Marine Biological Genetics and Breeding, No. J2021003; Central Public-interest Scientific Institution Basal Research Fund, CAFS, No. 2020TD71]
S912
A
1000-3096(2022)06-0070-10
10.11759/hykx20211119001
2021-11-19;
2022-04-06
国家自然科学基金项目(41806148); 浙江省近岸水域生物资源开发与保护重点实验室、温州市海洋生物遗传育种重点实验室联合开放基金(J2021003); 中国水产科学研究院基本科研业务费(2020TD71)
邢丽红(1981—), 女, 山东青岛人, 副研究员, 硕士, 主要从事水产品质量与安全研究, E-mail: xinglh@ysfri.ac.cn; 李兆新(1967—),通信作者, 研究员, E-mail: lizx@ysfri.ac.cn
(本文编辑: 谭雪静)
