基于高通量测序构建碘难治性分化型甲状腺癌ceRNA调控网络
2022-07-27罗莹莹卢其腾韦智晓
罗莹莹,卢其腾,韦智晓
(广西医科大学第一附属医院核医学科,广西 南宁 530021)
0 引言
甲状腺癌(Thyroid cancer,TC)是最常见的内分泌恶性肿瘤,占所有癌症的3.1%[1]。目前,手术+131I+TSH抑制治疗是分化型甲状腺癌(Differentiated thyroid cancer,DTC)的主要治疗手段,据SEER数据库显示美国甲状腺癌患者的5年生存率高达98.3%[2]。然而,约5%的患者应用131I治疗的效果较差,导致患者死亡,这部分患者的肿瘤细胞形态和功能发生退行性改变,浓聚碘的能力丧失,称之为碘难治性分化型甲状腺癌[3]。一项长期研究结果显示,远处转移的DTC患者对碘显著摄取的10年生存率约为56%,而没有显著碘摄取能力的患者10年生存率仅为10%[4]。因此,挖掘早期诊断RAIR-DTC的生物学标志物,明确RAIR-DTC的潜在发病机制,寻找新的治疗靶点,具有重要的临床意义。
长链非编码RNA (Long noncoding RNAs, lncRNA)是一类长度超过200 nt的RNA转录本,可干扰mRNA的剪切,直接结合蛋白调节其活性或者改变蛋白定位,以及作为ceRNA参与重组基因的表达,在转录、转录后调控和翻译等多个水平上调控基因表达[5]。2011年Salmena等[6]提出ceRNA假说,认为lncRNA可以通过miRNA应答元件(MRES)吸附miRNA,抑制miRNA发挥作用,进而间接调控靶mRNA的表达。多项研究表明,lncRNA在肿瘤发生发展及转移中发挥着重要的调控作用,Feng等人[7]发现lncRNAn384546通过作为miR-145-5p的竞争性内源性RNA调控AKT3来促进甲状腺乳头状癌的进展和转移;而Guo Q等人[8]研究发现lncRNA AB074169通过靶向KHSRP介导的p21表达抑制PTC肿瘤细胞增殖。然而,lncRNA及ceRNA调控网络在RAIR-DTC的研究鲜有报道。因此,本研究充分利用高通量测序数据,筛选RAIR-DTC中lncRNA和mRNA的差异表达谱,构建lncRN-miRNA-mRNA的ceRNA网络,探索lncRNA在RAIR-DTC中潜在的调控机制。
1 材料与方法
1.1 参与者和样本收集
本研究共收集在2013年至2019年在广西医科大学第一附属医院行131I治疗的72例患者,纳入标准:①经病理确诊为DTC;②至少接受2次131I治疗。排除标准:①抗甲状腺球蛋白抗体阳性;②放化疗史;③其他恶性肿瘤病史。根据影像学和血清学检查[3]分为RAIR-DTC组37例和NRAIR-DTC组35例,其中RAIR-DTC:①转移灶在清甲成功后的首次131I治疗后全身显像中即表现为不摄碘,致其无法从后续的131I治疗中获益。②原本摄碘的功能性转移灶经131I治疗后逐渐丧失摄碘能力。③部分转移灶摄碘,而部分转移灶不摄碘。④摄碘转移灶在经过多次131I治疗后虽然保持摄碘能力但仍在1年内出现病情进展。NRAIR-DTC的判断标准包括治愈和好转。分别从两组中随机选取3例患者的血液样本(3例RAIRDTC组和3例NRAIR-DTC组),分离血液中的单核细胞送至华大基因组学研究所进行RNA测序。本研究参与者均签署知情同意书,并获得了广西医科大学第一附属医院伦理委员会批准。
1.2 总RNA提取和lncRNA测序
根据制造商的说明,使用总RNA血液提取试剂盒(离心式,BioTeke公司)从全血中提取RNA,使 用Agilent Bioanalyzer 2100 (Agilent Technologies)测定RNA样品的浓度和纯度,并使用100 ng RNA进行1.5%琼脂糖凝胶电泳以进行初步质量控制。RNA样品满足以下标准:浓度>80ng/uL,RIN≥7以及A260/280值在1.9和2.2之间。琼脂糖凝胶电泳的完整性要求清晰可见的28S/18S条带无明显降解。
将采集到的细胞进行cDNA文库构建和RNA测序。使用DNBSEQ平台进行lncRNA测序,并将高质量读数与人类参考基因组(GCF_000001405.39_GRCh38.p13)进行比对。通过期望最大化(RSEM),将基因表达标准化为每百万映射读数(FPKM)每千碱基外显子模型的片段。
1.3 DElncRNAs和DEmRNAs的筛选
根据Michael I, et al.[9]中描述的方法,通过DEseq2软件分析并鉴定差异表达基因(DEGs),对编码蛋白的lncRNA和mRNA进行差异分析,设置|log2FC|>1,Qvalue<0.05,并对存在差异的lncRNA和mRNA绘制差异热图、火山图。
1.4 lncRNA、miRNA、mRNA互作预测
根据筛选出的lncRNA,应用mir-code数据库筛选与差异lncRNA相互作用的miRNA,再从miRTarBase、TargetScan、miRDB这3个miRNA靶基因预测数据库中取3个数据库均能预测到的靶基因,与步骤1.3所获得的差异mRNA取交集。基于上述筛选的lncRNA、miRNA、mRNA,整合得到lncRNA-miRNA-mRNA互作网络,并导入Cytoscape(版本:v3.9.0)绘制ceRNA网络图。使用Cytoscape核心插件cytohubba筛选degree数最高的10个靶基因,最终选取5个关键基因进行后续分析。
1.5 靶基因GO功能及KEGG信号通路分析
利用DAVID数据库对差异基因进行GO功能和KECG通路富集分析,研究RAIR-DTC的主要生物功能和信号通路,Q<0.05代表富集结果显著。利用R语言绘制GO和 KEGG富集分析气泡图。
2 结果
2.1 一般资料及RNA提取
随机收集3例RAIR-DTC患者和3例NRAIRDTC患者的全血样本进行lncRNA测序分析,女4例,男2例,平均年龄(42.83±15.20)岁。6份送检样本的提取结果情况如表1。
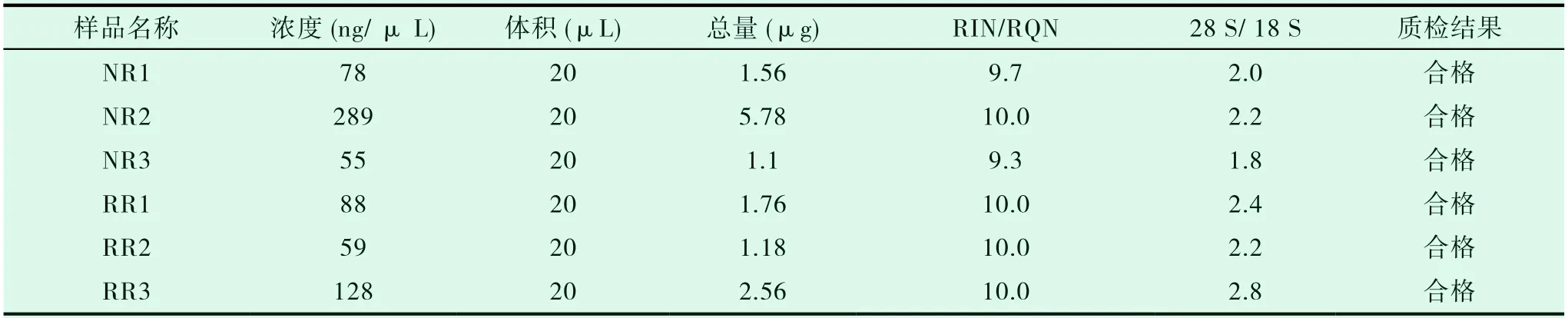
表1 RNA提取结果表
2.2 DEGs的鉴定
本研究在RAIR-DTC和NRAIR-DTC之间共鉴定出21个DEGs,上调12个,下调9个。共包括14个差异表达的lncRNA,上调9个,下调8个,其中有6个已知的lncRNA,8个新发现待注释的lncRNA;共有7个差异表达的mRNA,上调3个,下调4个(图1、表2、表3)。
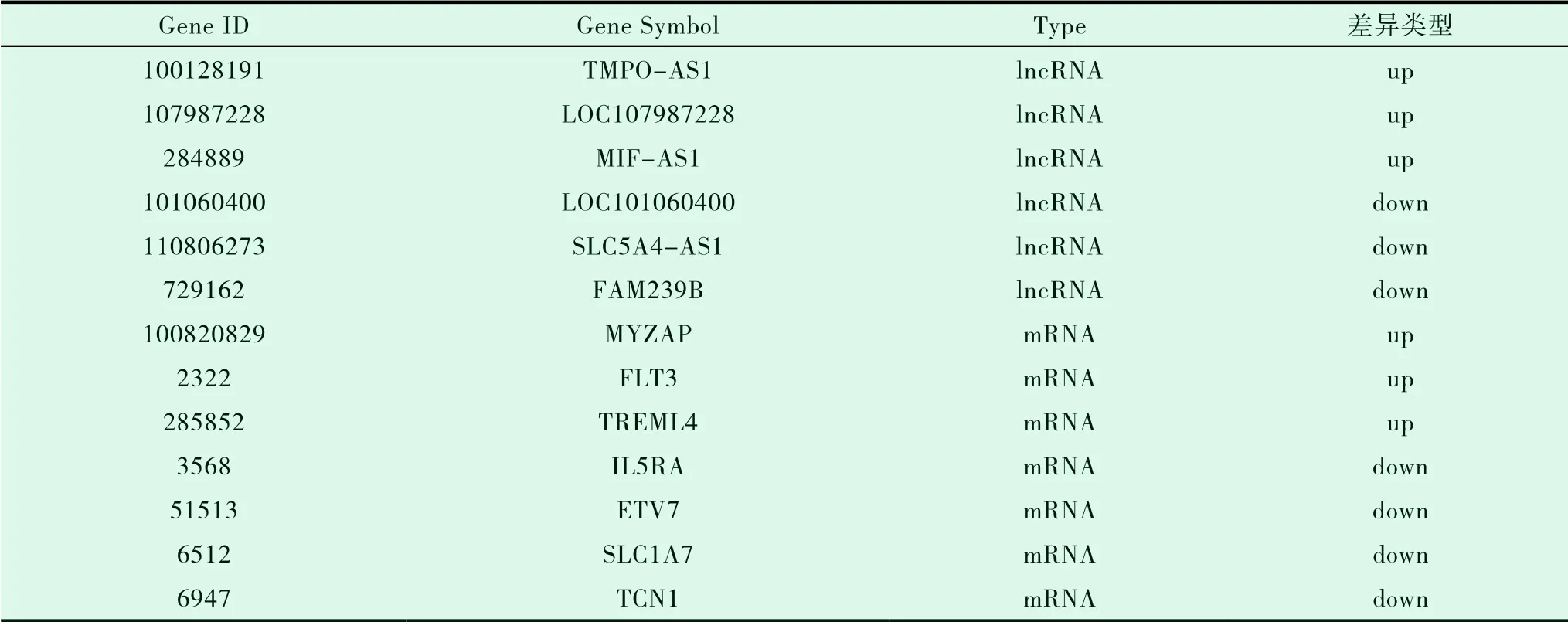
表2 已知差异基因(lncRNA、mRNA)汇总表
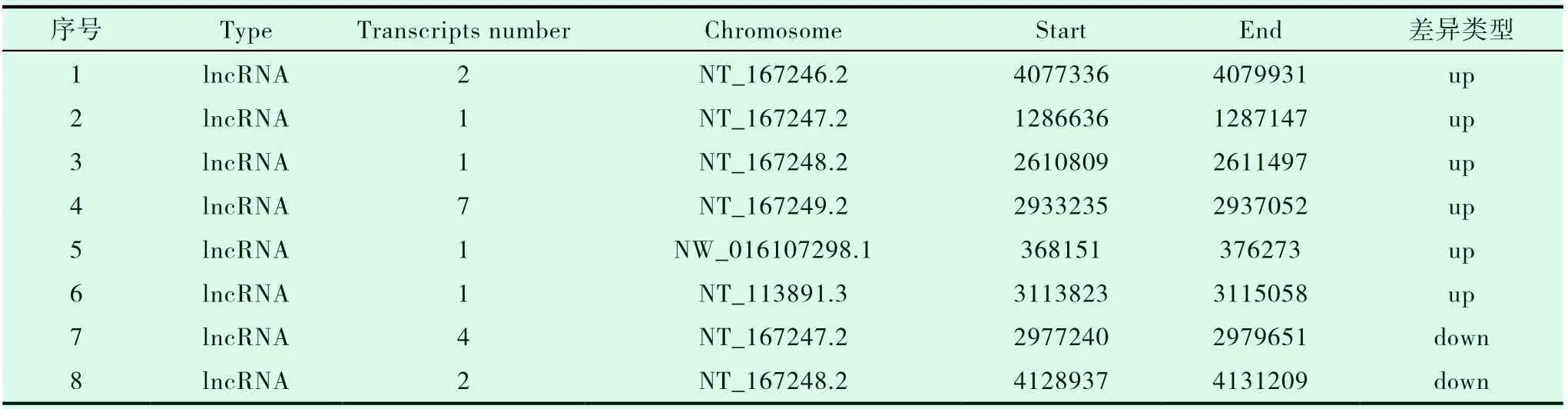
表3 新lncRNA汇总表
2.3 ceRNA网络
利用mircode等数据库共预测到168个miRNA和3079个mRNA,将预测到的mRNA与7个差异mRNA取交集,再删除不在该互作关系中的miRNA、lncRNA,整合后得到lncRNA-miRNAmRNA互作网络,绘制ceRNA网络(图2)。ceRNA调控网络中共包括174个节点(由5个lncRNA、168个miRNA、1个mRNA构建)和254条边,每条边表示lncRNA 与miRNA,miRNA与mRNA之间的关系。

图2 ceRNA调控网络
通 过Cytoscape中 的“cytohubba”插 件,选择“degree”top10的 基 因 作 为 关 键 基 因(图3),发现在关键度最高的5个lncRNA中,TMPOAS1、LOC107987228表达上调,其余3个lncRNA (LOC101060400、FAM239B、SLC5A4-AS1)均表达下调(图4)。通过ceRNA网络图分析lncRNA-miRNAmRNA之间的交互关系(表4),发现其中4个关键lncRNA (LOC101060400、FAM239B、SLC5A4-AS1、TMPO-AS1)与同一个miRNA(hsa-miR-1587)共同竞争靶mRNA(SLC1A7)的结合位点。

表4 ceRNA调控网络机制
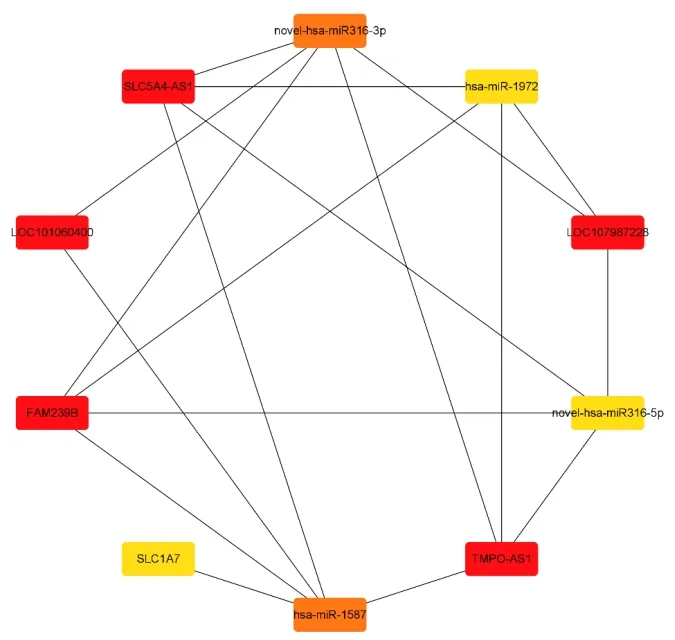
图3 鉴定到的关键基因
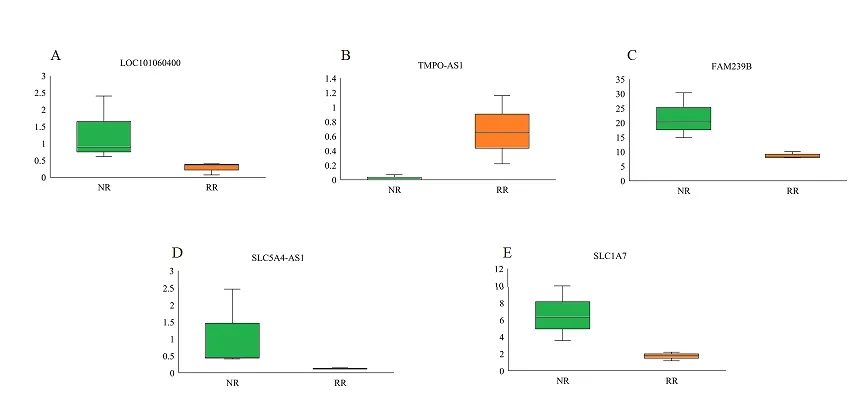
图4 关键基因在RAIR-DTC的表达水平
2.4 DEGs的GO功能和KEGG信号通路分析
本研究共获得30个富集的GO_CC、26个富集的GO_MF、63个富集的GO_BP及14条KEGG信号通路(图5)。GO分析结果显示:在细胞组成(cellular composition,CC)中,靶基因主要在白细胞介素-5受体复合物、RNA聚合酶II、全酶受体复合体富集(图5A);在分子功能(molecular function,MF)中,靶基因主要富集在细胞因子受体活性、白细胞介素-5受体活性、血管内皮生长因子活性(图5B);在生物过程(biological process,BP)中,靶基因主要在白细胞稳态、骨髓祖细胞分化、pro-B细胞分化、白细胞分化富集(图5C)。KEGG通路富集分析结果显示,靶基因主要富集在造血细胞谱系、癌症中的转录失调等信号通路(图5D)。
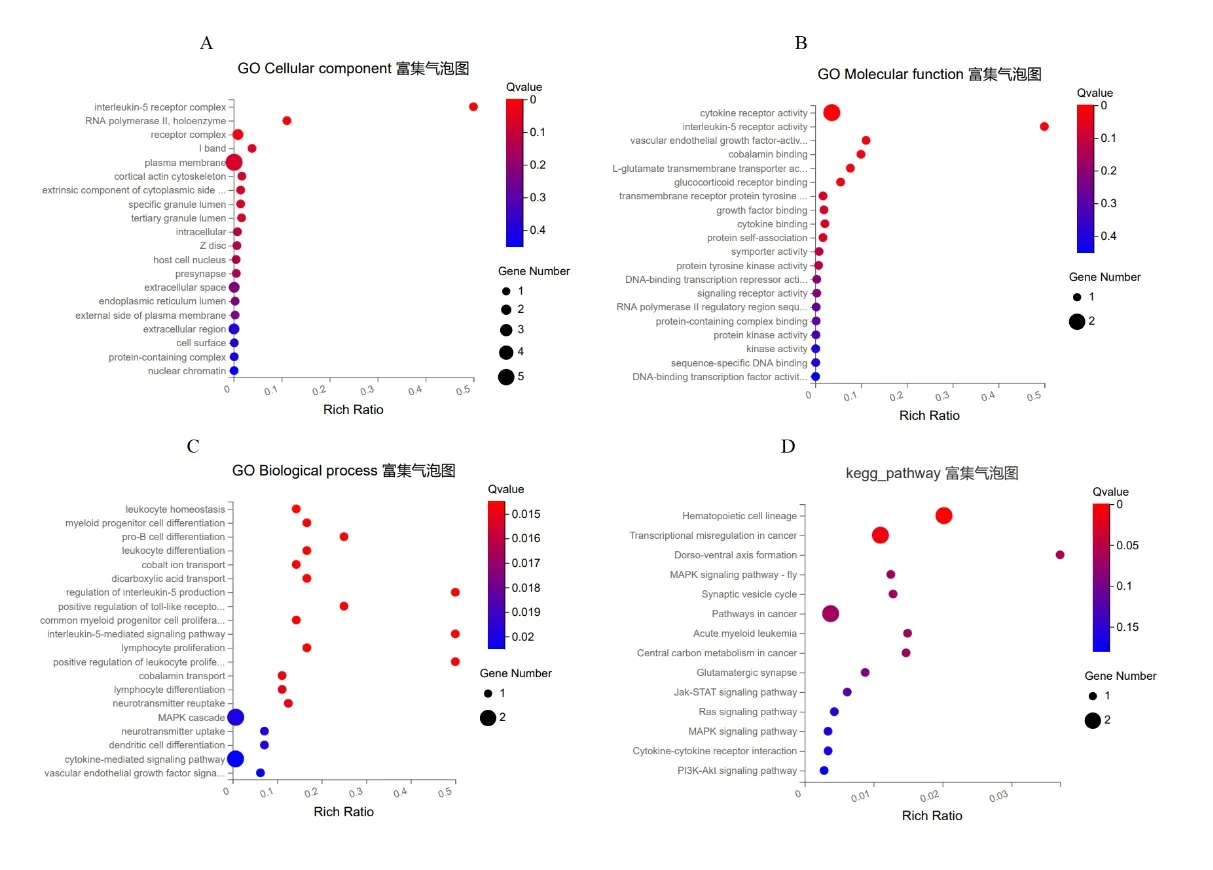
图5 靶基因的GO和KEGG富集分析
3 讨论
lncRNA在细胞的正常生理和病理过程中发挥着重要作用,越来越多的研究表明,lncRNA的失调与甲状腺癌的发病机制密切相关,如lncRNANEAT1、lncRNA XIST、lncRNA SLC26A4-AS1等[10-12]。miRNA是一类高度保守的内源性非编码小RNA,已发现多种miRNA与甲状腺癌的发生发展密切相关,如miRNA-21、miRNA-214、miR-98-5p等[10,13]。lncRNA相关的ceRNA调控网络在肿瘤进展中发挥重要作用,已经在胃癌、乳腺癌等多种肿瘤中对ceRNA网络进行了系统分析,目前对RAIR-DTC中lncRNA相关的ceRNA调控网络缺乏研究,其在RAIR-DTC中的表达模式和机制有待进一步探索。因此,本研究通过分析RAIR-DTC非编码RNA高通量测序数据,在RAIR-DTC中共鉴
定了特异性表达的14个lncRNA和7个mRNA,基于ceRNA假说成功构建了由5个lncRNA、168个miRNA、1个mRNA组成的ceRNA调控网络,进一步筛选出5个关键lncRNA,并通过GO和KEGG通路分析预测了RAIR-DTC中差异表达基因的功能,为RAIR-DTC临床诊断和治疗提供新靶标。
本研究结果显示,在ceRNA调控网络的5个关键lncRNA中,除TMPO-AS1、LOC107987228外,其他差异表达的lncRNA均下调。TMPO-AS1作为新发现的lncRNA,其功能是致癌的lncRNA,许多研究证实其在各种人类恶性肿瘤中异常高表达并与预后不良相关,如肺癌、肝细胞癌、甲状腺癌等[14-15]。Li Z等[16]研究结果表明TMPO-AS1在甲状腺癌组织和细胞系中高表达,并且TMPO-AS1通过海绵miR-498调节TMPO促进甲状腺癌的细胞增殖,我们的研究也同样发现TMPO-AS1在RAIR-DTC中高表达,因此,其可能在RAIR-DTC中发挥重要的生物学作用。而另外四个lncRNA(LOC107987228、LOC101060400、FAM239B、SLC5A4-AS1)此前均未报道与甲状腺癌相关,且与其他癌症的研究也未见报道,虽然这四个lncRNA在我们的研究中首次被报道为RAIR-DTC的潜在生物标志物,但相关证据仍不充分,未来需要进行深入研究。
miRNA是一类由内源基因编码的长度约为22 个核苷酸的非编码单链RNA分子,它通过与靶基因的非转录区结合调控靶基因的表达,进而影响细胞的生化过程和信号转导通路,导致肿瘤的发生和发展。在我们的研究中,发现miR-1587在RAIRDTC的调控中发挥着重要作用。miR-1587是近年来发现的一种富含G序列的新型miRNA,在其他疾病中有相关的研究。Tu D等[17]的研究揭示了miR-1587 / IRF7轴介导了M2巨噬细胞诱导的乳腺癌进展。Liu R等[18]研究发现,miR-1587过表达增强了DNA双链断裂的形成,阻止了结直肠癌(CRC)细胞的生长,并通过直接抑制LIG4表达增强了CRC细胞的放射敏感性,揭示了miR-1587是CRC放射治疗的潜在靶标,并确定miR-1587是一种有效的电离辐射(IR)敏化剂。miR-1587在DTC/RAIR-DTC中的研究有限,值得我们进一步研究。
在参与ceRNA调控网络的基因中,KEGG分析显示,这些基因主要富集在两条通路中:造血细胞谱系通路和癌症中的转录失调信号通路。Sun T团队[19]经大样本生信分析研究发现,癌症中的转录失调信号通路在PTC发病机制中发挥重要功能的途径。Zhao L等[20]的最新研究表明,在甲状腺癌中异常甲基化的DEGs主要与癌症中的转录失调、MAPK信号通路和TC中的内在凋亡信号通路有关。在本研究中确定的ceRNA调控网络为进一步研究提供了有用的线索。
在本研究中,我们构建了lncRNA-miRNAmRNA的ceRNA网络,鉴定了与RAIR-DTC相关的差异表达lncRNA和mRNA,其中一些RNA(如TMPO-AS1)可能代表RAIR-DTC临床诊断的新型候选生物标志物,但需要进一步验证和研究。此外,我们还需要进一步的功能实验研究,以确定ceRNA网络的组成部分在RAIR-DTC中的具体功能作用。
