8例囊性纤维化患儿临床及基因突变分析
2022-07-25张娜刘建华褚亚娟帅金凤黄坤玲
张娜 刘建华 褚亚娟 帅金凤 黄坤玲
(1.河北医科大学研究生学院,河北石家庄 050017;2.河北省儿童医院呼吸二科,河北石家庄 050031)
囊性纤维化(cystic fibrosis,CF)是一种常染色体隐性遗传的慢性致死性疾病,发病机制为囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)突变导致CFTR通道蛋白的离子和水转运障碍,累及呼吸道、消化、生殖多个系统,临床表现为反复呼吸道感染、胰腺功能不全、男性双侧输精管缺失等[1]。CF 发病率存在种族、地域差异性,多见于白种人。美国发病率约1∶4 000[2],英国约1∶2 500[3],日本约1∶350 000[4],我国少见,暂无该病的流行病学资料。近几年随着医学技术进步,我国CF 患者被更多地确诊,但存在诊断延迟现象。本文总结分析8 例CF 患儿的临床及遗传学特征,让更多临床医生了解并重视该病,减少误诊漏诊。
1 资料与方法
1.1 研究对象
以河北省儿童医院2018~2021年确诊的8例CF患儿为研究对象,进行回顾性病例总结分析。
CF的诊断标准[5]:(1)一项或多项典型CF临床特征;(2)CF家族史;(3)新生儿CF筛查试验阳 性;(4) 汗 液 氯 离 子 浓 度≥60 mmol/L;(5)CFTR2个致病变异(父母双方等位基因各提供1个);(6)CFTR 功能障碍(鼻电位差和/或肠道电流测量)。满足其中(1)~(3)中至少1条,加上(4)~(6)中至少1条即可诊断为CF。
胰腺CF的诊断标准[6]:符合CF的患儿具有胰腺受损的临床表现,主要为胰腺外分泌功能不全(pancreatic insufficiency,PI)[7]导致的脂肪、蛋白质和糖类的吸收障碍,可表现为腹泻、体重下降,严重时可引起脂肪泻、营养不良及生长发育迟缓等症状。影像学检查可见胰腺受损征象,如胰腺饱满、胰腺实质回声不均、胰腺脂肪化、胰腺纤维化、胰腺扩张、胰腺萎缩、胰腺囊肿样变等。
1.2 方法
病例资料收集:通过医院电子病历系统,收集患儿的确诊年龄、起病年龄、性别、既往史、个人史、家族史、体格检查、各项检查结果、治疗及随访情况等。
CFTR基因变异分析:(1)突变筛查:高通量测 序(二 代 测 序, next‐generation sequencing,NGS) 检测变异,多重连接探针扩增技术(multiple ligation probe amplification,MLPA)检测基因缺失与重复及拷贝数变异。(2)基因数据分析:变异分级依据美国医学遗传学与基因组学学会 (American College of Medical Genetics and Genomics,ACMG)制定的2015测序变异的解读指南进行致病性等级分类,查询CFTR2 数据库(https://www. cftr2. org/)、CFTR‐France 数 据 库(https://cftr.iurc.montp.inserm.fr/cgi‐bin/home.cgi#)行致病性分析。蛋白功能预测采用REVEL、SIFT、PolyPhen‐2、 MutationTaster、 GERP+ 软 件 。(3)Sanger 测序、qPCR 测序进行验证。以上均由北京迈基诺基因科技有限公司、智因东方北京全谱医学检验实验室执行。
1.3 统计学分析
采用描述性分析。计量资料采用范围(中位数)表示,计数资料采用例数表示。
2 结果
2.1 一般资料及临床特征
8 例CF 患儿中,男5 例,女3 例,无CF 家族史,父母非近亲结婚,无白种人通婚史。诊断年龄为3~48个月(中位年龄8个月),发病年龄为0~24个月(中位年龄2.5个月)。8例患儿的临床特征见表1。临床表现为反复呼吸道感染7 例,鼻窦炎3例,支气管扩张4例,腹泻8例,脂肪泻3例,可疑PI 6 例,胰腺CF 1 例,营养不良5 例,假性Batter 综 合 征(Pseudo‐Bartter syndrome,PBS) 4例。2例(例3、例6)行汗液电导率检测,显示汗液电导率增高。患儿最常见的呼吸道感染病原为铜绿假单胞菌(Pseudomonas aeruginosa,PA;4例)。7 例行胸部CT 检查,其中4 例存在支气管扩张,见图1。5 例行支气管镜检查及治疗,均可见栓状、条索状分泌物,见图2。
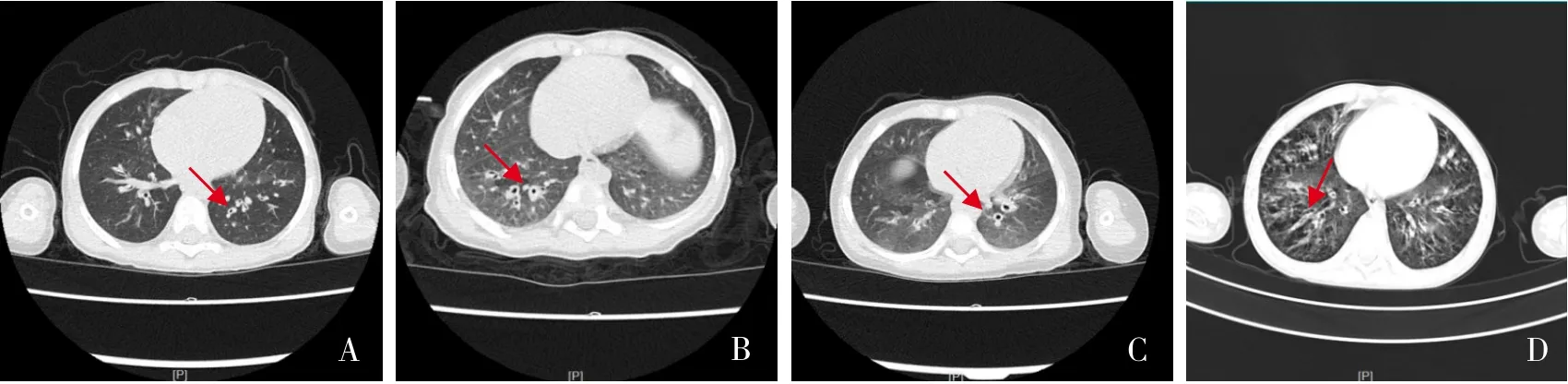
图1 4例患儿胸部CT表现 A:例3,双肺炎症伴小囊腔,左肺上叶支气管起始部狭窄。双侧胸膜局部增厚,可见支气管壁增厚、支气管扩张(箭头所示)。B:例4,双肺炎症,右肺下叶支气管壁增厚、扩张(箭头所示)。C:例6,双侧支气管肺炎,小气道病变不除外;右肺上叶支气管起始部、右肺中间支气管管腔细,可见支气管壁增厚、支气管扩张(箭头所示)。D:例7,双侧支气管扩张(箭头所示)伴双肺炎症。
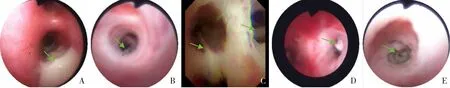
图2 5例患儿支气管镜下表现 A、B、C、D、E分别为例3、例4、例5、例6、例7,均可见栓状、条索状分泌物(箭头所示)。
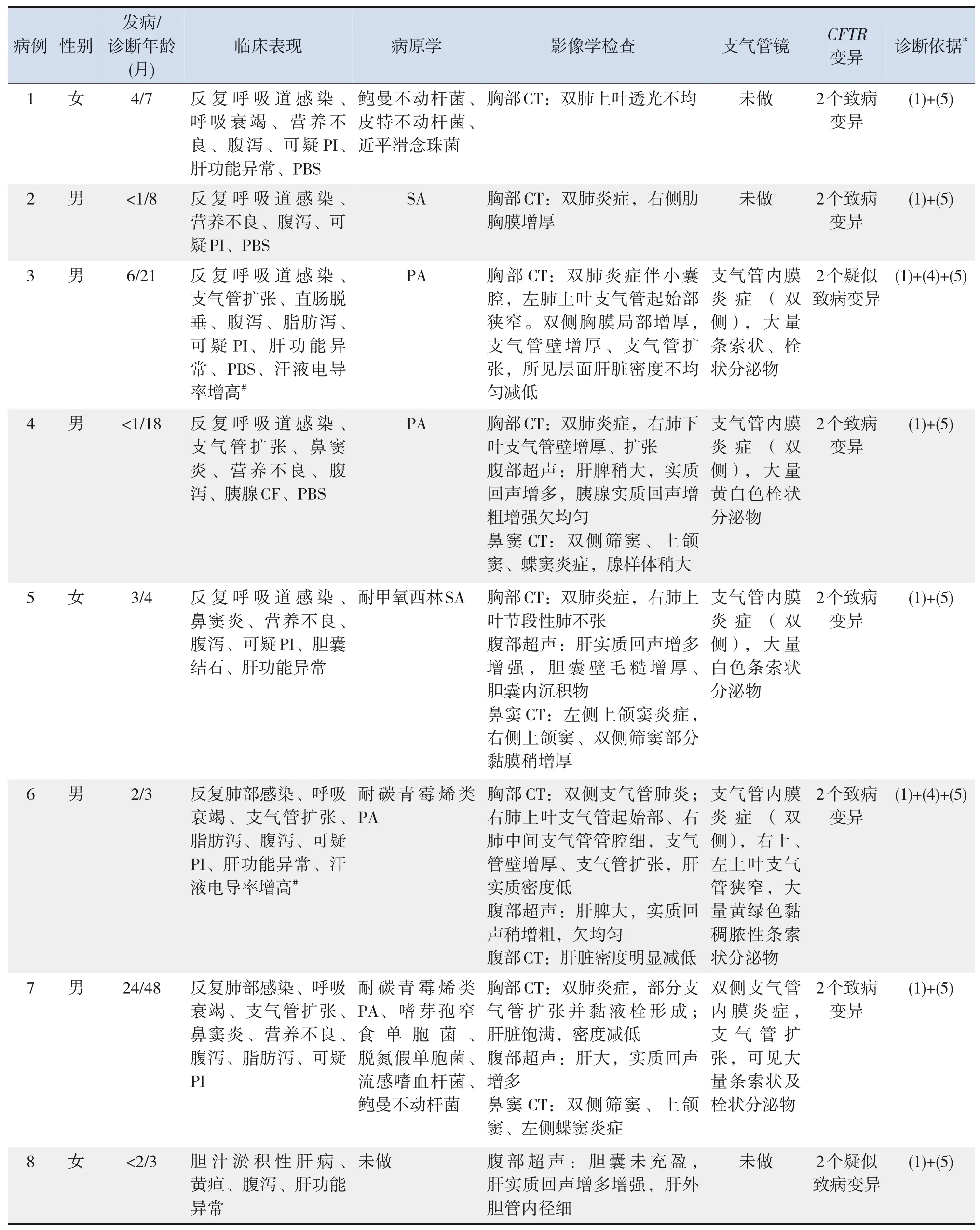
表1 8例CF患儿临床表现、辅助检查及诊断依据
例1、例2、例3、例4入院时行电解质及血气分析,发现存在电解质紊乱及代谢性碱中毒,见表2。
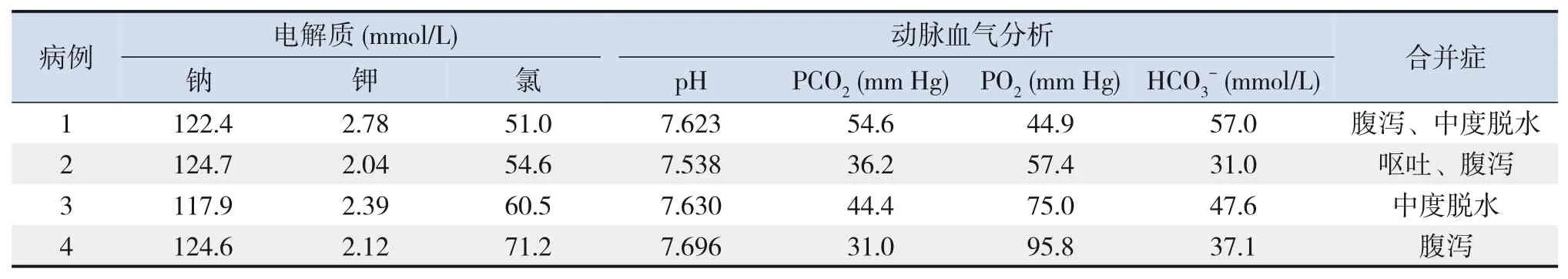
表2 4例CF患儿血电解质及血气分析结果
2.2 基因检测结果
8 例患儿均行基因检测,均检出CFTR基因突变。例2 为纯合突变,其余7 例为复合杂合突变。共发现16个变异位点(其中4个为新发现变异),包括移码突变5个,无义突变4个,错义突变4个,外显子缺失2个,剪接突变1个。最常见的基因突变类型是p.G970D(3 例),观察到1 例F508del 基因突变。8 例CF 患儿的基因突变位点及致病性分析见表3。
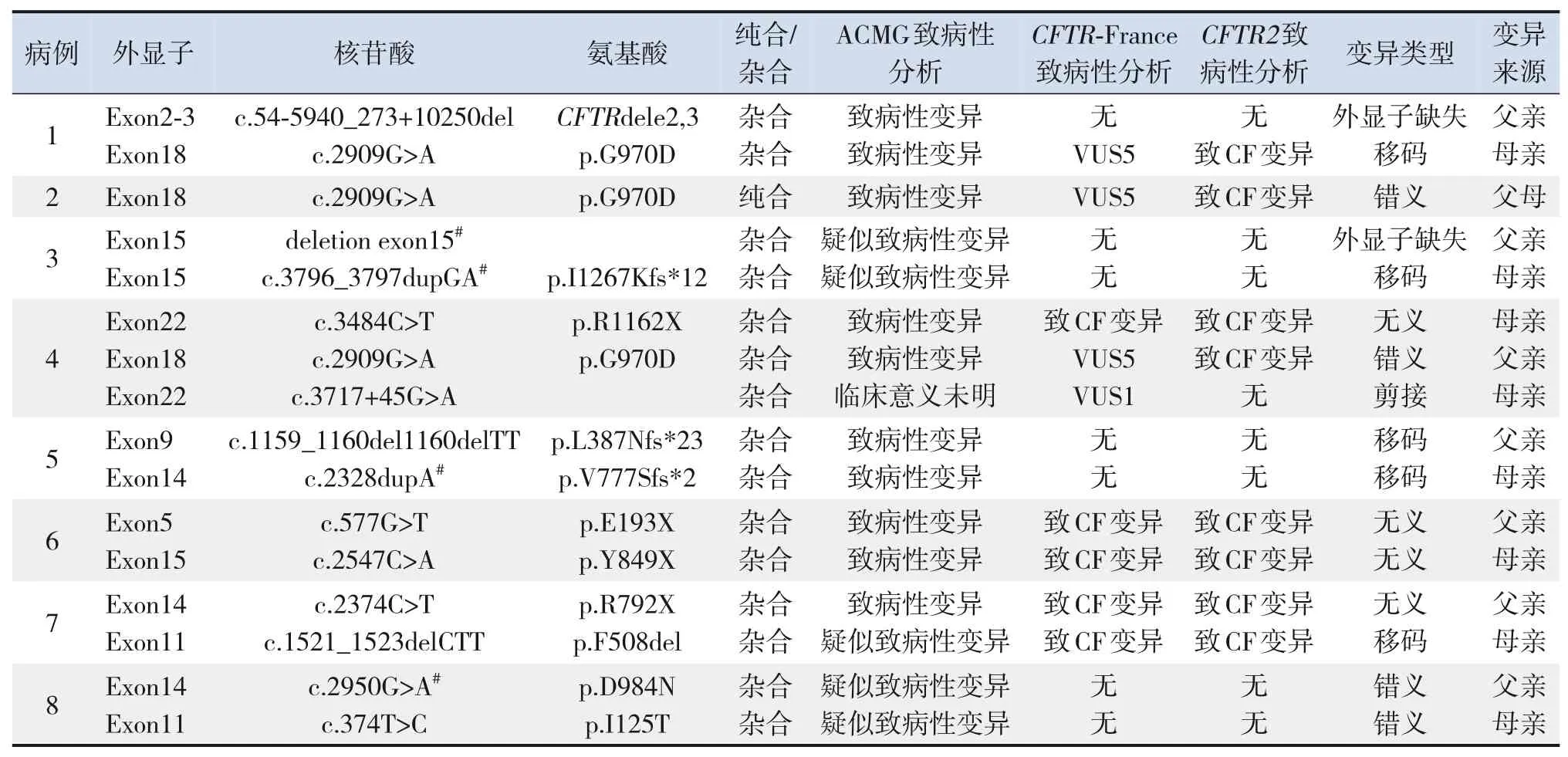
表3 8例CF患儿基因突变位点及致病性分析
3 讨论
CF 是一种多见于白种人的常染色体隐性遗传疾病,预后差、病死率高。1989年Riordan等[9]首次发现CF 的致病基因位于7q31,致病机制是CFTR基因隐性突变致CFTR 通道蛋白活性减低或丧失引起疾病。CFTR 是一种cAMP 依赖调控的氯离子通道蛋白,是三磷酸腺苷结合盒转运体家族的一员。CFTR蛋白广泛存在于气道、胃肠道、生殖道等多种器官的上皮组织顶膜,调节离子和水的转运。CFTR通道蛋白的离子和水转运障碍,黏液增加,管腔堵塞,引起多系统功能障碍。
CF 患者通常以反复呼吸道感染为首发表现[10],常表现为鼻窦炎、鼻息肉、反复肺部感染、支气管扩张、气胸、喘息、咯血、呼吸衰竭、杵状指等。CFTR功能障碍,气道上皮细胞Cl-分泌减少而上皮细胞钠离子通道吸收Na+增加,造成细胞内高渗,从而使得气管表面液体减少,气道内黏液增加,纤毛摆动受限、清除能力下降,细菌定植引起反复炎症,肺结构破坏,肺功能进行性下降,最终患者呼吸衰竭造死亡[11]。本研究中7 例CF 患儿临床表现为反复肺部感染,其中伴有鼻窦炎3 例,支气管扩张4 例,呼吸衰竭3 例,例7 反复肺部感染,出院3个月后因呼吸衰竭死亡。7 例患儿均存在肺部影像学异常表现,其中4例存在支气管扩张。胸部CT是检测CF患儿肺部情况的重要检查。有研究表明,影像学比肺功能更能敏感地反映肺部严重程度,CF典型胸部CT表现包括支气管扩张、黏液堵塞、空气滞留、灌注异常和肺气肿[12]。临床医师需定期监测患者肺部情况,了解疾病进展,对于反复呼吸道感染患儿应警惕CF。
本研究中6 例患儿诊断为可疑PI,因存在CF基础疾病,有腹泻、脂肪泻、营养不良及生长发育迟缓等症状,粪便常规可见大量脂肪球,影像学检查可见胰腺回声改变,口服胰酶症状可缓解,但我院不能进行胰腺功能检测,故最终不能确诊。CFTR功能障碍时,黏稠液体阻塞胰腺管道,导致炎症、脂肪变、纤维化,随着年龄增长胰腺功能失代偿,PI比例逐渐升高。国外报道约90%CF患者合并PI[13],我国学者报道14.1% CF 患者出现PI[14]。我国CF患者合并PI比例明显低于国外,可能与我国CF 患者基因突变类型多为轻型有关,故临床表现轻;也可能与胰腺功能检测困难,大部分医院不能完成该检测至最终确诊病例少有关。随着诊断技术的推广与提高,PI 确诊率可能有所上升。
国外报道CF 相关性肝病(cystic fibrosis‐related liver disease,CFLD)为CF 患者继呼吸衰竭、肺移植并发症的第三大死亡原因,约10%患者进展为肝硬化,2%~3%进展为门脉高压[15],目前国内相关报道数量少。CF 患者因黏稠且具有肝毒性胆汁阻塞胆道,早期可仅表现为肝脏氨基转移酶升高,但随病情进展可能会出现胆汁淤积性肝硬化、门静脉高压、晚期肝病。本研究中例1、例3、例5、例6、例8存在肝功能异常,影像学示肝脾、胆囊改变,但国外研究显示肝功能异常、黄疸、胆汁淤积、胆囊结石并不是CFLD 危险因素,男性、小年龄、PI、严重CFTR突变等是CFLD 的危险因素[16-17],但是该5 例患儿仍需警惕CFLD,临床医师对存在高危因素的患儿应提高警惕以早期识别CFLD。
例1、例2、例3、例4 婴儿期均有PBS 且为首发或主要表现。PBS发病机制为CFTR介导的Cl-和上皮细胞钠离子通道介导的Na+吸收减少,大量Na+和Cl-经汗液丢失,引起机体肾素-血管紧张素-醛固酮系统激活,造成电解质紊乱。国内外报道PBS 可为CF 的首发表现,出现在疾病早期,且容易复发[18-19]。这提醒临床医师,具有PBS表现的儿童需警惕CF,注意监测患儿电解质情况,尤其是天气炎热季节。
本研究8 例患儿中有4 例为PA 定植感染,其中2 例为耐碳青霉烯类PA。PA 属于机会致病菌,具有分泌毒素、形成生物膜、群体感应等特点[20]。2019年美国CF基因会数据显示CF肺部病原菌43%为PA,25%为耐甲氧西林的金黄色葡萄球菌,14%为非结核分枝杆菌,12%为甲氧西林敏感的金黄色葡萄球菌[2]。美国CF 基金会指南强烈建议首次感染PA患者应行早期根除治疗[21]。我国目前针对PA感染主要是依据药敏试验选择敏感抗生素抗感染治疗,根除PA治疗临床少见。
目前已有2 000 多种CFTR基因突变类型[10]。国外研究报道,该基因最常见的突变类型是cDNA的508 位点苯丙氨酸缺失(p.F508del),90%的CF患者中发现至少有1个F508del 等位基因,属于第Ⅱ类突变,CFTR 蛋白功能严重丧失,多伴有PI[10,22]。目前我国仅报道3 例携带F508del 突变患者[23-25]。Tian等[25]报道我国CF突变类型多为少见突变,最常见的突变类型为p.G970D。本研究最常见的突变类型亦为p.G970D,共检出3例,F508del突变1 例,与ACMG/美国妇产科医师协会(American College of Obstetricians and Gynecologists,ACOG)[26]提议的23个突变筛查模板不同,与国外报 道[27]的CF 最 常 见 的5 种 突 变c. 1521_1523delCTT、c.1624G>T、c.1652G>A、c.350G>A、c.3909C>G 不同,提示我国与国外CF 基因突变谱不同。本研究发现2个基因大重排,例1患儿行全基因组测序发现CFTRdel2,3突变,例3患儿全外显子基因检测只检测到1个基因突变位点,进一步行MLPA 检 测 发现deletion exon15 突变。2018 年美 国CF 基因会病人登记信息示约3%患者检测出<2个致病性变异[28]。这提醒临床医师疑诊CF患者若只检测出1个基因突变位点,可进一步行全基因组测序及MLPA 检测。本研究发现deletion exon15、c. 3796_3797dupGA(p. I1267Kfs*12)、 c. 2328dupA(p.V777Sfs*2)、c.2950G>A(p.D984N)在人类基因突变数据库中未收录,为新发现的变异,这扩大了我国CF的基因突变谱。
汗液氯离子水平升高是CF 患者主要临床表现及诊断条件之一。我国研究报道,汗液电导率检测可用来代替汗液氯离子水平检测进行CF诊断,当汗液电导率超过80 mmol/L,考虑CF诊断[8]。本研究中2 例患儿在北京儿童医院行汗液电导率检测,汗液电导率分别为110 mmol/L、140 mmol/L,均高于80 mmol/L,支持CF诊断。
本研究中8例患儿诊断中位年龄为8个月,中位发病年龄为2.5个月,提示CF患者存在诊断延迟现象,考虑以下原因:(1)我国CF 患儿临床表现不典型,以肺部疾病为主要表现,合并PI比例低,诊断困难;(2)我国CF 诊断试验不完善,尚未开展新生儿CF 筛查、鼻腔电位差、肠道电流测量,开展汗液氯离子/电导率检测的医疗机构少,基因检测因价格昂贵致应用受限;(3)在我国CF 属于罕见病,临床医师对该疾病认识不足,造成误诊漏诊。
综上,我国CF存在诊断延迟现象。我国CF患儿临床表现不典型,最常见的临床表现为反复呼吸道感染,合并PI 比例明显低于国外。临床上如果发现患儿存在反复呼吸道感染(支气管扩张、鼻窦炎等),合并或不合并消化系统表现和PBS,呼吸道病原学检测示PA 阳性,需警惕CF。p.G970D 为CF 患儿最常见的突变类型,F508del 少见,与国外CF基因突变谱不同。本研究为CF未来精准治疗、制定我国基因突变谱、研究基因突变谱与临床表型关系、产前诊断提供了参考依据。
