儿童进行性家族性肝内胆汁淤积症3 型一例并文献复习
2022-07-25郭淑仪欧榕琼周敦华张碧红王海燕檀卫平
郭淑仪 欧榕琼 周敦华 张碧红 王海燕 檀卫平
进行性家族性肝内胆汁淤积症3 型(PFIC3)是ATP 结合盒转运蛋白A4(ABCB4)基因突变引起的遗传性胆汁淤积症。ABCB4 基因位于染色体7q21.1 上,包含28 个外显子,其中27 个外显子含编码序列,参与编码多耐糖蛋白(MRD3)。MRD3 分布在肝脏、B 淋巴细胞、心脏和肌肉等,主要位于肝毛细胆管膜。MRD3 的功能是分泌磷脂酰胆碱并将其转运至胆汁。MRD3 表达水平和(或)功能异常可影响磷脂酰胆碱的分泌和转运,引起胆石症、肝内胆汁淤积症。PFIC3 临床表现为肝脾肿大、黄疸,数年内可进展为肝硬化等。目前已报道的PFIC3 相关的ABCB4 基因致病性突变约有150 多种,已有研究多数为个案报道。现报道本院收治的1 例PFIC3 儿童病例,并检索国内外的相关文献进行分析,以提高临床医师对PFIC3的认识水平。
对象与方法
一、收集1 例PFIC3 儿童病例的病历资料
本院2020 年6 月28 日收治1 例PFIC3 患儿,收集并分析其病史、体格检查、实验室及辅助检查、治疗及转归等资料。
二、文献检索
以“儿童”及“进行性家族性肝内胆汁淤积症3 型”(包括中英文)为检索词,对以下数据库截至2022 年1 月收录的文献进行检索:PubMed、中国生物医学文献服务系统(SinoMed)、CNKI、万方数据知识服务平台、维普中文科技期刊数据库,收集并分析检索到的PFIC3 患儿的临床及遗传学特点。
结 果
一、1 例PFIC3 患儿的病历资料
1.入院主诉及检查
患儿男,1 岁3 个月,因B 超发现肝肿大5 d于2020 年6 月28 日入院。患儿母亲为孕产,患儿足月顺产,出生后无黄疸病史,生长发育与同龄儿相仿。其母亲于2008 年被检出华支睾吸虫抗体阳性,未予治疗。其胞兄(1 岁10 个月)于2008 年因“腹胀1 年余、身目黄染3 个月”住院,肝功能检查提示胆汁淤积性肝炎,华支睾吸虫抗体阳性,被诊断为“①华支睾吸虫病(中华);②缺铁性贫血;③支气管肺炎; ④EB 病毒感染”,予驱虫、抗感染治疗后出院,2015 年(8 岁)因肝硬化、慢性肝衰竭、肝性脑病、消化道出血、肺出血、脓毒血症死亡。患儿父母非近亲结婚,父母、2 个胞姐(15 岁、6 岁)及1 个胞兄(4 岁6个月)均身体健康,否认家族其他成员有肝病病史,否认有其他特殊病史。
体格检查:发育正常,营养尚可。身目稍黄染,未见肝掌、蜘蛛痣。双侧下颌角后方及颌下可触及数枚绿豆大小淋巴结,均质软、界清,无压痛,与周围组织无粘连。心、肺无异常。腹部未见腹壁静脉曲张,全腹质软,未扪及包块,无压痛、反跳痛,肝肋下9 cm 可触及、质软、边锐,脾肋下未触及。移动性浊音(-),肠鸣音正常。双下肢无水肿。
实验室及辅助检查:血红蛋白129 g/L,白细胞14.09×10/L,淋巴细胞7.77×10/L,嗜酸性粒细胞0.78×10/L ,血小板391×10/L。尿胆红素(+),尿比重1.009。粪便未见异常。AST 179 U/L,ALT 170 U/L,总胆红素 46.1μmol/L,直接胆红素26.2 μmol/L(正常值范围0.0~3.4 μmol/L),间接胆红素19.9 μmol/L(3.4~18.8 μmol/L),甘油三酯3.80 mmol/L,白蛋白38.5 g/L,球蛋白 40.5 g/L,γ-谷氨酰转移酶(GGT) 137 U/L,碱性磷酸酶278 U/L,总胆汁酸 203.2 μmol/L。EB 病毒壳抗体-IgG(+),EB 病毒壳抗体-IgM(-),EB 病毒早期抗体-IgG(-),EB 病毒核抗体-IgG(-), EB病毒壳抗体-IgG(高亲和力)(-), 壳抗原IgG 亲和力指数22%,EB 病毒 4.24×10copies/mL。凝血酶原时间12 s,部分凝血活酶时间41.5 s (延长),纤维蛋白原2.43 g/L。巨细胞病毒DNA 定量、嗜肝病毒抗体、单纯疱疹病毒抗体、风疹病毒抗体、弓形虫抗体、血粪肝吸虫、血吸虫、肺吸虫抗体未见异常。乳酸、血糖、酮体、血尿氨基酸及有机酸、血铜蓝蛋白、血氨未见异常。血红蛋白电泳、红细胞渗透脆性试验、葡萄糖6 磷酸脱氢酶活性、IgA、IgG、IgM、补体C3、补体C4、RF、CRP 未见异常。腹部B 超、MRI 提示肝脾肿大。腹部磁共振血管造影、磁共振胰胆管成像未见明显异常。心电图、泌尿系统超声检查、心脏彩色多普勒超声检查(彩超)未见异常。因患儿家属原因未行肝活组织检查(活检)。患儿母亲复查华支睾吸虫抗体阴性。患儿父母肝功能正常。本研究获中山大学孙逸仙纪念医院伦理委员会批准(批件号:SYSKY-2022-107-01),患儿父母均签署了知情同意书。
2.基因突变检测
对患儿及其父母的基因进行高通量测序。基因检测(北京迈基诺基因科技股份有限公司)结果如下:在ABCB4 基因上发现1 个纯合突变,突变位置的染色体位置为chr7:87037402,转录本外显子NM_000443;exon25,核苷酸氨基酸c.3230C>T(p.T1077M),为错义突变,见表1。根据美国医学遗传学与基因组学学会(ACMG)指南初步判定该突变临床意义未明。经Sanger 测序进行家系验证分析,该位点突变来源于父母,患儿父亲该位点为杂合突变,患儿母亲该位点为杂合突变,见图1。由于经济原因,家族中其他成员未进行基因检测。
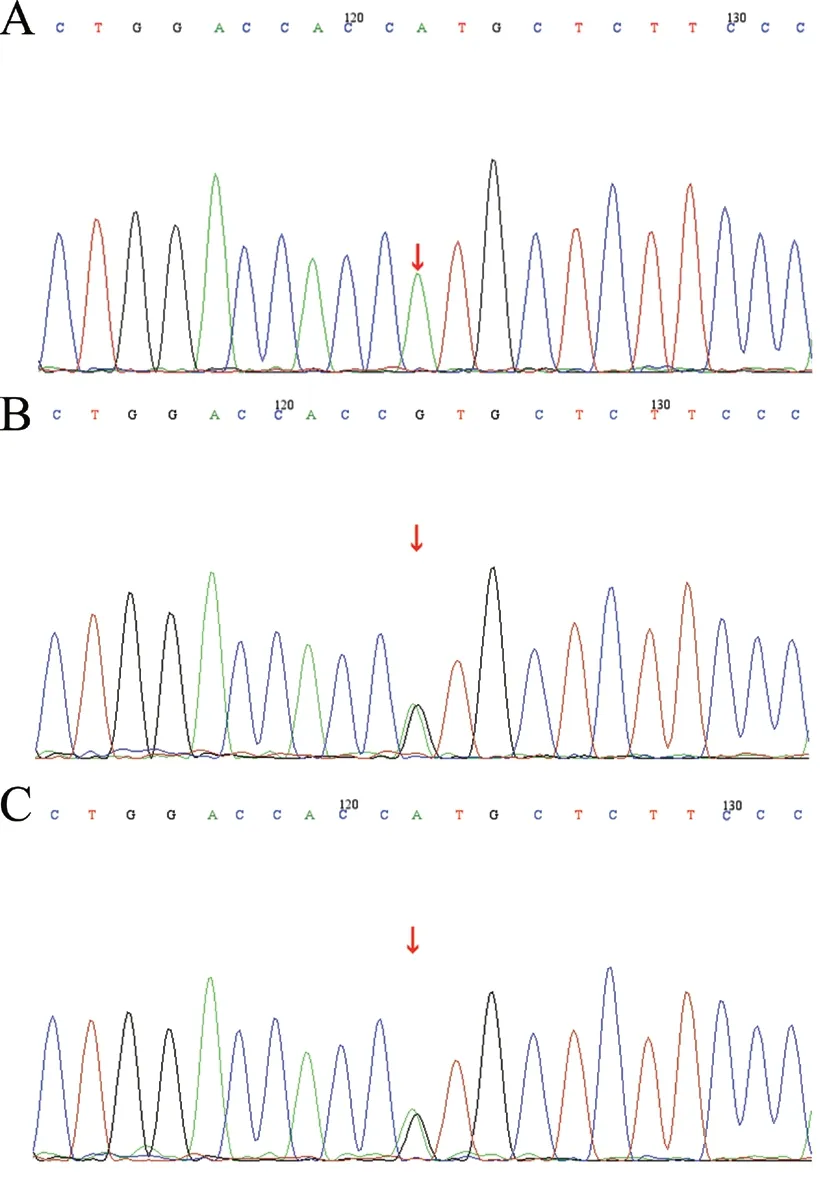
图1 一例PFIC3 患儿及其父母的ABCB4 基因Sanger测序图
生物信息学分析显示,ABCB4 基因第25 外显子c.3230C > T 突变导致其编码的MRD3 蛋白第1077 号苏氨酸被替换为蛋氨酸。这一突变在正常人群数据库中的频率为0.0000199,为低频突变,因此为纯合稀有突变。采用SIFT、PolyPhen_2、Mutation Taster、PROVEAN 预测均提示该突变为有害,见表1。Genome 系统发现ABCB4 基因在从低等单细胞生物到灵长类动物的胆盐输出泵蛋白(BSEP)同源肽中高度保守,提示该位置的氨基酸改变可能影响蛋白的功能。经PubMed BLAST 系统分析突变所影响的蛋白结构域,采用SWISS-PDB Viewer 4.1.0 软件预测发现,第1077 号氨基酸周围肽链空间结构明显改变,见图2A、B。

图2 PubMed BLAST 系统分析一例PFIC3 患儿ABCB4 基因突变所影响的蛋白结构域

表1 错义突变预测结果
3.治疗与转归
结合患儿病史、各项检查结果及基因突变情况,诊断其为PFIC3、EB 病毒感染,予更昔洛韦抗EB 病毒治疗2 周,复方甘草酸苷护肝、注射用丁二磺酸腺苷蛋氨酸(思美泰)、熊去氧胆酸(UDCA)利胆等对症治疗2 周后复查肝功能示AST、ALT 及胆红素较前下降,胆汁酸下降不明显。患儿出院后长期口服复方甘草酸苷、UDCA。随访至2021 年6 月仍存活,但AST、ALT、胆红素、胆汁酸仍偏高。
二、PFIC3 儿童病例的相关文献检索结果
收集到已报道的PFIC3 儿童病例共11 例,见表2。已报道的PFIC3 儿童病例首发症状多为黄疸、皮肤瘙痒、肝脾肿大、肝功能异常(以GGT升高为主)。所用保守治疗一般为UDCA 护肝治疗,大部分患儿肝酶虽然下降但疾病仍持续进展,最终需肝移植。目前暂未发现ABCB4 基因 c.3230C >T 纯合错义突变的报道。

表2 PFIC3 儿童病例的相关文献检索结果

序号 第一作者 基因突变位点及蛋白1基因突变位点及蛋白2 MDR3蛋白表达发病年龄,性别,首发症状等ALT(U/L)GGT(U/L)总胆红素病理治疗结局9 Giovannoni I[4]p.R595X p.R595X 无描述4 岁,女无描述无描述无描述胆汁性肝硬化、门静脉周围纤维化逐渐进展为肝硬化7 岁接受肝移植,目前9岁,已治愈10 Park H J[5] c.749G > C p.A250P c.857C > T p.A286V c.1069T > C p.F357L c.1091C > T p.A364V c.1424T > C p.V475A c.2144C > T p.T715I c.2210C > T p.A737V c.3577G > A p.A1193T A364V、A737V和A119 3MRD3活性降低无描述无描述无描述无描述无描述无描述无描述11.1 陈友惠[6] c.431G>A p.Arg144G In无描述未检测4 岁,男,腹胀,肝脾肿大,有家族史12221896 未检测UCDA 治疗 随访3 个月,肝功能好转11.2 陈友惠[6] c.431G>A p.Arg144G In无描述未检测11 岁,女,肝功能异常17121177 肝小叶结构紊乱、肝细胞水样变性、单个核细胞增生活跃UCDA 治疗 随访3 个月,肝功能好转12 李爱芹[7] c.2212A >T c.1694C > G c.2570C >T 无描述4 岁,男,肝功能异常111126 无描述肝小叶结构紊乱、假小叶形成、肝细胞弥漫性水样变性UCDA 治疗 随访3 年疾病无进展,但仍有皮肤瘙痒13邓梅[8] c.1006-2A >G c.3580C > T 未检测4 岁,男,皮肤黄染17344150 无描述UCDA 治疗 失访,结局不详
讨 论
根据北美及欧洲胃肠道协助组共识,婴幼儿期发病原因不明的胆汁淤积性肝病1/3 为先天性胆道闭锁等结构异常所致,1/3 为遗传代谢性疾病,其他因素包括感染、肿瘤等。本例患儿母亲及胞兄曾被检出华支睾吸虫抗体阳性,但前者无肝功能异常,患儿多次行寄生虫相关检查未见异常,故考虑华支睾吸虫感染可能性小。患儿存在EB 病毒感染,但EB 病毒引起的肝功能受损多以胆红素、转氨酶升高为主,较少影响胆汁酸排泄,因此EB 病毒感染不足以解释患儿出现的所有临床症状。结合患儿临床症状、各项检查结果以及家族史,排除肝结构异常、占位性病变、感染等情况,考虑患儿为PFIC 的可能性大。目前PFIC 尚无明确的诊断标准,确诊依赖于基因检测。
PFIC 是一种常染色体隐性遗传病,由于基因突变导致胆汁酸形成及分泌异常引发一系列临床症状,表现为婴幼儿期出现进行性的黄疸和瘙痒、不同程度的生长发育障碍、脂溶性维生素缺乏导致的维生素K 缺乏性出血等。大多数患者在青春期前进展为肝硬化、肝衰竭。血液生化检查特征为血清胆汁酸和转氨酶水平升高,可伴血清胆红素及碱性磷酸酶水平升高。根据基因突变类型,PFIC 被分为1~6 型。PFIC1 由ATP8B1 基因突变所致,该基因编码胆汁淤积相关蛋白-1(FIC1),患者大部分在1 岁前发病,部分可到青春期才出现胆汁淤积症状,症状较轻的基因突变可表现为良性复发性肝内胆汁淤积症1 型(BRIC1)。PFIC2由ABCB11 基因突变所致,该基因编码BSEP,由于突变类型的差异,临床表现差异大,重症者常在新生儿期发病,进展迅速,在10 岁前进展为肝衰竭,轻症者则表现为BRIC2。PFIC4 由紧密连接蛋白2(TJP2)基因突变所致,TJP2 分布于全身各处,单独的基因突变除会引发肝脏疾病外还会引发耳聋、神经和呼吸系统等肝外症状。PFIC5 由NR1H4 基因突变引起,该基因编码法尼酯X 受体(FXR),此类患儿多在新生儿期发病并迅速发展为终末期肝病,伴有严重的维生素K 非依赖性障碍及甲胎蛋白水平异常升高,是最严重且预后最差的PFIC。PFIC6 由肌球蛋白Vb(MYO5B)基因突变引起,该基因编码MYO5B 蛋白,与肝细胞、肠上皮细胞密切相关,因此常合并难治性腹泻。
PFIC3 由ABCB4 基因突变所致,基因位于染色体7q21 上,主要编码MRD3,可分为4 个区:2个同源的核苷酸结合区,分别由3 个外显子编码(外显子12、13、14 和外显子25、26、27),能够结合和水解ATP;还有2 个同源的跨膜区。MRD3 主要在肝毛细胆管膜上表达,其功能是分泌磷脂酰胆碱乳化胆盐和胆固醇。MRD3 功能异常会导致胆汁内缺乏磷脂,不能乳化胆盐和胆固醇,进一步引起胆管损伤、胆石沉积等。PFIC3 不同于其他基因引起的PFIC 类型,ABCB4 基因突变导致的MRD3 缺失并不影响胆盐的分泌和排泄功能,而会使更多的可溶性GGT 反流入血,导致血清GGT 水平升高,其他PFIC 类型患者的血清GGT 正常或大致正常。回顾本例患儿情况,患儿及其胞兄在婴幼儿期发病,表现为胆汁淤积性肝炎的症状,包括肝肿大以及转氨酶、血清胆红素、碱性磷酸酶、胆汁酸、GGT 水平升高,无伴其他肝外表现,提示PFIC3 可能。
研究显示,ABCB4 基因突变与肝脏相关性疾病密切相关,常见的ABCB4 基因缺陷可致PFIC3、低磷脂相关性胆石症综合征、妊娠期高GGT 肝内胆汁淤积症、慢性胆管病、成人肝硬化及肝胆恶性肿瘤等。根据碱基置换突变可将基因突变分为同义突变、错义突变、无义突变、终止密码突变。人类基因库Human Genome Mu(http://www.hgmd.org/)显示与ABCB4 基因相关的疾病有150多种,已发现与PFIC3 有关的基因突变有50 余种。已有文献报道致病基因不同、同一致病基因的不同基因突变会引发不同的临床表现且病情严重程度也不同。其中PFIC3 纯合无义突变病情重。笔者通过基因检测及Sanger 测序发现本例患儿存在ABCB4 基因c.3230C > T (p.T1077M) 纯合错义突变,突变来源于其父母,国内外尚无此突变的文献报道。采用蛋白功能损伤预测软件预测此突变为有害。患儿胞兄病程进展迅速,未能进行基因检测明确诊断,其他家族成员亦未进行基因检测完善家系分析,故仍需长期追踪其他家族成员是否出现肝脏相关性疾病。
目前尚无针对PFIC3 病因学的治疗方案,一般采用以改善肝功能、缓解皮肤瘙痒、补充维生素改善营养状况为主的治疗方案,以及治疗肝硬化、DIC 等远期并发症。UDCA[10~20 mg/(kg· d)]可改善50% PFIC3 患者的生物化学指标异常情况,对于PFIC3 错义突变的患者效果最佳,但对基因无义突变或错义突变引起MRD3 完全缺失的患者无效。利福平、考来烯胺可缓解皮肤瘙痒症状。补充脂溶性维生素、中链甘油三酯有助于改善患者的营养状态。肝移植是治愈PFIC3 的唯一方法,移植指征包括严重皮肤瘙痒、明显生长发育迟缓、肝硬化及肝衰竭。有研究者报道了5 例接受肝移植的PFIC 患儿,其中3 例为PFIC1、1 例为PFIC2、1 例为PFIC3,PFIC3 患儿于移植后第19日肝功能即恢复正常,随访1 年未出现并发症。
本文报道的PFIC3 患儿有明确的遗传病史,伴血清高GGT,经基因诊断明确为PFIC3,基因突变c.3230C > T(p.T1077M)的发现扩展了ABCB4基因突变谱,为诊断PFIC3 提供依据。本例患儿未能行肝活检检测MRD3 的水平,家系中其他成员亦未进行基因测序及长期随访等,仍需进一步的长期随访。
