原位光沉积法制备Au/TiO2室温催化氧化CO的研究
2022-07-20李求忠戴文新黄仁昆颜桂炀
李求忠,戴文新,黄仁昆,颜桂炀*
(1.宁德师范学院 化学与材料学院,福建 宁德 352100;2.福州大学 能源与环境光催化国家重点实验室,福建 福州 350108)
众所周知,CO 会使人体中毒,甚至威胁生命.在质子交换膜燃料电池(PEMFC)中,氢气主要来源于水煤气变换,因水煤气变换制取氢产生的少量CO(0.5%~2.0%)同样会造成电极上贵金属(Pt 或Pt-Ru)中毒而失活[1-2],氢气在进入燃料电池之前必须进行CO的脱除净化处理.因此,在富氢的反应气中,CO的优先氧化也是非常重要的研究任务.
近几十年来,关于CO 的催化转化一直是催化研究领域的基础课题,是兼具理论研究和应用价值的体系.自Haruta等[3]在还原性载体上负载超细金纳米粒子实现了低温催化氧化CO以来,原本一直被认为是惰性的金受到了极大的关注,而其中Au/TiO2在CO 催化氧化中比Pt/MO 具有更优越的活性而成为研究热点之一[4-10].CO 分子在金纳米粒子上吸附活化(Au-CO),与金-载体界面的晶格氧发生反应生成CO2,造成晶格氧的缺位(氧空位的生成),而氧分子在缺陷位上吸附活化并补充晶格氧,使晶格氧得到再生[11-12],因此,O2在界面的吸附活化成为提高其活性的关键因素之一.对于如何提高Au 负载型催化剂的效率,认为关键在于Au纳米粒子大小、分散状态的控制和对CO 的吸附活化能力[13],往往Au颗粒越小和分散度越高,其活性也会越高[14],当Au 的粒径小于3 nm 时,催化剂表现出了良好的性能,而当其粒径大于5 nm时,则会失去其独特的催化性能[15].
超细Au纳米颗粒由于小尺寸效应和表面效应使其具有较低的熔点,研究表明,2 nm Au纳米粒子的熔点只有300°C左右[16],当热处理温度超过300°C时,则会容易导致其烧结而聚集,破坏Au粒子原有的不对称配位,致使其催化剂活性明显下降.因此,要想获得高度分散且稳定的Au 纳米粒子,需要合适的制备方法和对条件的严格控制.传统浸渍法[17]负载Au粒子通常粒径较大(>10 nm),分散度较差,Au与载体间作用也较弱.此外,已报道的方法还有共沉淀法[18]、沉积-沉淀法[19-20]、化学蒸发沉积法[21]和光沉积法[22].用光沉积法在TiO2上负载Au,相对于沉积-沉淀法可以获得更小的Au纳米颗粒[23].Chen 等[24]在P25上用光沉积法负载Au,获得粒径在1.5 nm 左右的Au 纳米粒子,而且发现Au 的粒径会随着光沉积时间的延长而增大,制备的催化剂用于CO氧化具有较好的催化性能和选择性.
本课题用溶剂热法制备了钛乙二醇前驱体,通过原位光沉积法成功在其表面负载Au 纳米颗粒,经过进一步热处理后,获得的Au/TiO2催化剂在室温下可高效催化氧化CO,表现出很高的活性和可见光下的光促作用.通过TPD、in situDRIFTS 和in situESR 探讨了Au/TiO2催化剂对CO 和O2的吸附活化与转化行为.
1 实验部分
1.1 材料制备
1.1.1 TiO2(EG)和TiO2的制备 将2.5 mL 钛酸四丁酯溶于75 mL 乙二醇(EG),在磁力搅拌下将2.5 mL水滴入溶液中.将所得白色溶胶转移到内衬聚四氟乙烯的不锈钢高压釜中,在150°C下恒温4 h,然后冷却至室温.离心收集白色产物,分别用水和乙醇洗涤数次.在80 °C 下干燥过夜后,将产物标记为TiO2(EG).将TiO2(EG)在马弗炉中350°C下煅烧3 h,得到纯TiO2.
1.1.2 Au/TiO2的制备 用原位光沉积法制备Au/TiO2.将一定量的HAuCl4溶液(0.01 g·mL-1)加入到100 mL水中,用0.1 mol·L-1NaOH 溶液将pH 值调至9.0.将TiO2(EG)(250 mg)加入其中并搅拌3 h得到悬浮液.用PLS-SXE300 型氙灯(北京普发科技有限公司)进行光沉积30 min,通过离心收集浅紫色的产物,并进一步用水洗涤数次,然后在80°C 下干燥12 h,产物标记为Au/TiO2(EG).将Au/TiO2(EG)放入马弗炉中,在350°C 下煅烧3 h,得到紫色的Au/TiO2,Au 负载的理论质量分数为0.6%.以上所有试剂为分析纯,均来至上海国药集团,水为高纯水.
1.2 材料结构表征
X 射线粉末衍射(XRD)在Bruker D8 Advance 上进行,使用Cu Kα辐射源(λ=0.154 18 nm).在SU8000型扫描电子显微镜(SEM)、TECNAI G2F20型透射电子显微镜(TEM)和JEM-ARM200F型球差校正的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)进行样品形貌分析.用物理化学吸附仪(Micromeritics Instrument ASAP2020)测定比表面积,并且通过BJH方法从吸附等温线的解吸分支计算孔体积和孔分布.
用Varian Cary 5000 UV-Vis-NIR 型光谱仪测试样品的紫外可见漫反射光谱(UV-Vis DRS).X 射线光电子能谱(XPS)在Thermo Scientific ESCALab250 光谱仪上测试,用Al KαX 射线束(1 486.6 eV)收集,用C1s 峰(BE=284.80 eV)校准.光电化学实验在Metrohm-Autolab AUT302N 型电化学工作站上进行,使用典型的三电极电化学池配置,填充0.2 mol·L-1Na2SO4电解质,铂片电极为对电极,Ag/AgCl(饱和KCl)电极为参比电极.使用Bruker A-300E型电子顺磁共振光谱仪在室温下记录原位电子自旋共振(in situESR)光谱信号,工作频率为9.84 GHz,功率为15.92 mW.CO 的程序升温脱附(TPD) 在Micromeritics Autochem2910 仪器上进行,实验方法参照本课题组前期的工作[25],在升温过程中同时记录m/z=28、44的质谱信号,分别用于检测CO、CO2分子.
原位漫反射红外傅里叶变换光谱(in situDRIFTS)是在带有MCT 检测器的Bruker Vexter 80v FT-IR型光谱仪上进行,首先,将样品装入in situDRIFTS 反应器池中,在He 气氛中(50 mL·min-1)200°C 下预处理1 h,降温至25°C后,采集样品的光谱作为采集背景.将体积分数为5%CO(平衡气为高纯He)作为探针气体以10 mL·min-1流速引入反应池,连续采集数据30 min.数据采集范围为4 000~1 000 cm-1,分辨率为8 cm-1,每个光谱扫描64 次,以降低信噪比.
1.3 催化剂活性测试
催化剂催化氧化CO 的活性在常压固定床流动反应器中进行[26].将催化剂样品(300 mg,粒径约为0.2~0.3 mm)装入方形石英池中,用冷却水系统控制反应温度为25 ℃,总流速为100 mL·min-1(WHSV=20 000 mL·h-1·gcat-1).反应混合气中CO 和O2体积分数均为0.3%,高纯He为平衡气.在石英反应器上用铝箔封闭进行暗条件下的测试.在引入可见光时,使用300 W 的氙灯作为光源,加载滤光截止片(<420 nm和>760 nm),保证可见光通过.使用气相色谱(Agilent 7890D,TDX-01填充柱,TCD 检测器)进行在线分析出口气体中CO、O2及CO2的浓度含量.CO转化率(XCO,%)计算公式如下:

2 实验结果与讨论
2.1 材料制备与形貌表征
用XRD 对TiO2(EG)、TiO2和Au/TiO2样品进行表征,可得到样品的晶相结构,结果如图1所示.在前驱体TiO2(EG)的XRD 谱图中没有观测到明显的特征衍射峰,说明为无定形态,而TiO2和Au/TiO2样品谱图中,分别在晶面(101)(004)(200)(105)(211)(204)(116)(220)和(215)检测到锐钛矿TiO2特征峰(JCPDS No.21-1272),较宽的衍射峰可以说明样品结晶度较差.此外,Au/TiO2样品中都没有检测到Au的衍射峰,这是因为Au在样品中呈高度分散状态.

图1 TiO2和Au/TiO2样品的XRD图
为了更直观地获得样品表面形貌及负载Au 粒子的分布形态,本研究对样品进行SEM、TEM、HRTEM 和STEM 测试.从图2 的SEM 图可以看出,TiO2(EG)和Au/TiO2样品均为不规则的粒状形态,负载Au 和热处理后的Au/TiO2样品颗粒变大.从图3(a)的TEM 图可以看到Au纳米粒子能均匀分散,从图3(b)的HRTME 图可以观测到晶面间距为0.35 nm 的晶格条纹,对应于锐钛矿型TiO2的(101)晶面,以及晶面间距为0.23 nm 的Au纳米粒子,归属为Au的(111)晶面.用球差校正高角度暗场扫描透射电镜对Au/TiO2样品进行测试,结果见图3(c),从图中可以看到Au纳米粒子主要负载在TiO2载体的边角处,Au通过与载体边界的强相互作用嵌在载体的表面,降低其表面自由能,从而更好地保持稳定存在,从高分辨STEM图可以清晰地看到Au纳米粒子中Au原子呈独特的排布特征,也可以看到在经过热处理后的样品Au纳米粒子没有严重烧结,仍有明显的边角和台阶,这种不对称的配位Au 原子将更有利于反应物分子的吸附和活化.根据STEM 结果统计的粒径分布情况可以看出(图3(d)),Au 纳米粒子的粒径主要分布在3.5 nm 左右,而且呈较好的正态分布,说明用原位光沉积法负载的Au 具有均匀的小纳米粒子和良好的分散度.

图2 (a)TiO2(EG)和(b)Au/TiO2样品的SEM图

图3 Au/TiO2样品的(a)TEM,(b)HRTEM,(c)STEM和(d)Au纳米粒子的粒径分布图
通过N2的物理吸脱附分析载体和催化剂的比表面积和孔径分布,图4给出了TiO2和Au/TiO2样品的物理吸脱附等温曲线.从图4可知,样品的吸脱附曲线中都有一滞后环,属于典型的IV型吸附曲线,表明样品为类介孔材料.从图5 样品孔径分布图和表1 物理吸附结果可看出,TiO2在负载Au 和经过热处理后,比表面和孔容略有减小,而孔径相应减小较为明显.

表1 TiO2和Au/TiO2样品的物理吸附结果

图4 (a)TiO2和(b)Au/TiO2样品的吸脱附等温曲线
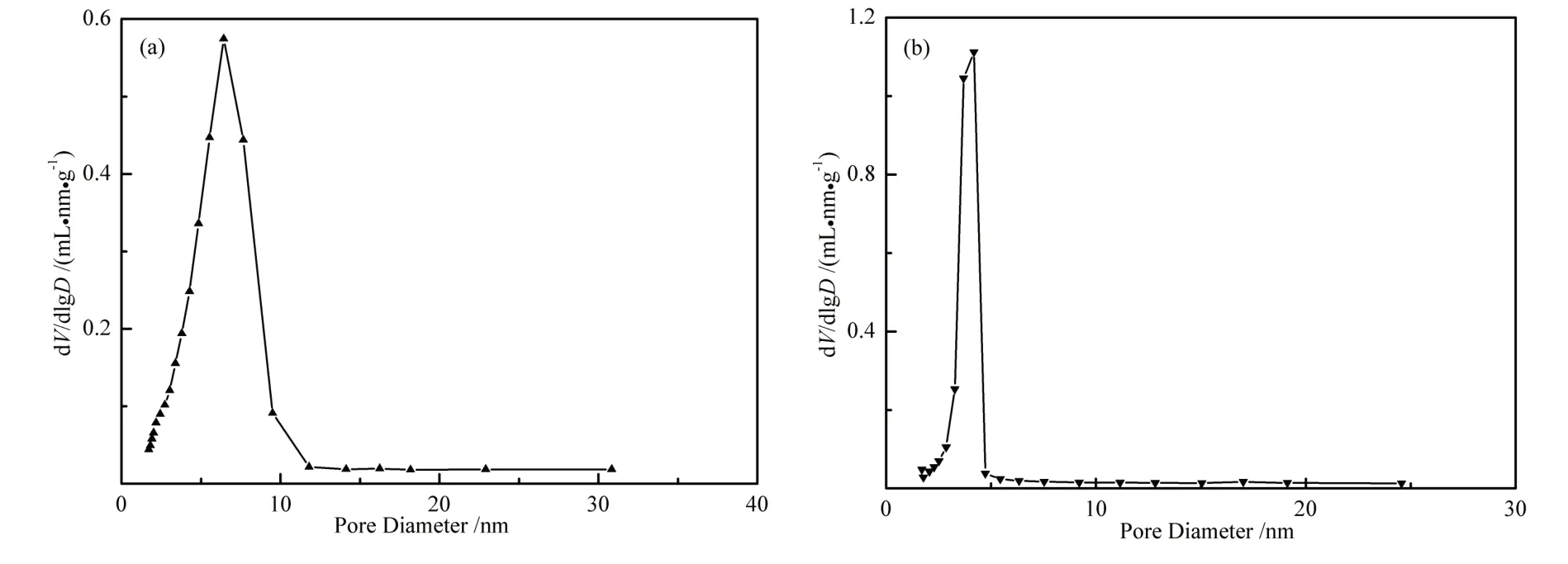
图5 (a)TiO2和(b)Au/TiO2样品的孔径分布图
对TiO2和Au/TiO2样品进行了紫外-可见漫反射表征,考察样品对可见光的吸收性能,结果见图6所示.从图6可看出,TiO2和Au/TiO2样品在紫外区都有TiO2的本征吸收峰,Au/TiO2样品在可见光区域(579 nm)出现了明显的吸收峰,归属于Au纳米粒子的局域表面等离子共振(LSPR)效应,这将促进Au纳米粒子表面电荷密度增加,同时也有利于CO在Au粒子表面的吸附和活化,从而更好地实现CO的催化氧化.

图6 TiO2和Au/TiO2样品的UV-Vis DRS图
图7 是TiO2和Au/TiO2样品在可见光下响应的瞬时光电流.TiO2本身由于表面缺陷引起了可见光激发而表现一个较弱的光电流,在负载Au后,可见光照下引起Au纳米粒子的LSPR效应,引起光电流的增加.从图中可发现Au/TiO2样品比TiO2的光电流增加表现得更为明显,说明其电荷迁移效果更高,这也表明了可见光引起的Au纳米粒子LSPR效应对Au和TiO2界面的电子转移起决定性作用.

图7 可见光下TiO2和Au/TiO2样品的瞬态光电流响应
图8为Au/TiO2(EG)和Au/TiO2的XPS谱图.从图8(a)可知,样品表面有吸附一定量的碳氢化合物和碳酸盐,C 元素的来源主要为催化剂制备过程中表面残留的乙二醇基、碳酸盐或空气中吸附的含碳物质,Au/TiO2(EG)样品在结合能为286.14 eV 处有很强的C1s峰,这主要是乙二醇基中的碳,在经过热处理后,该峰强度和峰面积比明显减弱.从图8(b)Au 4f 的XPS 光谱中发现,Au/TiO2(EG)样品没有探测到氧化态Au+4f7/2(85.00 eV)和Au3+4f7/2(86.60 eV)的特征峰,而只有Au0的特征峰,说明经过光还原后,Au已被顺利地还原出来.而且探测到的金属态Au 4f7/2(83.25 eV)和Au 4f5/2(86.90 eV)电子结合能低于标准的结合能(84.0和87.7 eV)[28],而热处理后的Au/TiO2样品Au 4f的电子结合能(83.04和86.69 eV)更是发生了负移,这一现象类似于其他研究者的结果.原因是TiO2的费米能级比Au纳米粒子的高,载体表面缺陷位的电子会从载体转移到Au纳米粒子的表面,形成新的费米能级.O 1s的XPS谱图见图8(c)所示.结合能位于529.99和529.58 eV的主峰归属于晶格氧Ti-O,位于531.06 eV的为表面吸附的羟基O-H[10,29],而结合能在532.03 eV的可归属于表面吸附氧(Oads),包括碳酸盐或羧酸盐有机物种[30],也有研究者将此结合能位置归为水[31]或O2在氧空位上吸附后形成的O-2[32].经过热处理后,乙二醇基和表面吸附物种的热分解导致羟基和吸附氧的特征峰明显减弱.从图8(d)的Ti 2p高分辨XPS谱图可以看到,Au/TiO2(EG)样品在电子结合能458.63和464.33 eV 处探测到的两个峰,Au/TiO2样品对应的峰负移到458.49 和464.20 eV,分别归属于Ti 2p3/2和Ti 2p1/2的自旋-轨道峰,双峰分裂差值为5.7 eV,与TiO2的标准谱(458.5 和464.2 eV)相吻合[33].样品经过热处理后,表面化学吸附的-OH和乙二醇基等物种分解脱附,形成更多的缺陷位,使得其表面具有更高的电子密度,促使Au 4f、Ti 2p和晶格氧的电子结合能都发生了不同程度的负移.

图8 Au/TiO2(EG)和Au/TiO2样品的XPS图
2.2 催化剂活性测试
为了考察Au/TiO2样品的稳定性和最大转化效率,分别进行了光暗交替实验,结果见图9所示.在cCO=0.3%时进行稳定性测试(图9(a)),结果发现,暗条件下,催化剂在连续120 h内转化率都达到100%,没有任何衰减迹象,表现出了良好的稳定性,说明在较低CO浓度下Au/TiO2样品对CO具有高效的催化氧化效率.随后在样品未经任何再生预处理的情况下,将反应气的CO浓度提高到3%,在初始的5 h样品的活性略有衰减,主要原因是在较高浓度的CO 反应气下,初始阶段生成的类碳酸盐等中间物种部分在样品表面累积,无法快速转化成气态的CO2而脱附,导致活性位点减少,随着反应的吸/脱附过程逐渐达到稳定,在催化剂表面的催化氧化反应达到一种动态平衡[34,35].Au/TiO2样品在暗条件下CO转化率达到80%以上,转化频率(TOF)可达650 h-1以上,引入可见光后,转化率更是超过98%,TOF值进一步提高到860 h-1,效率提高了约37%,而且与暗条件下相比显得更为稳定,表现出良好的光助活性和很好的光助稳定性.此样品活性较常规化学还原法制得Au/TiO2的活性(光暗条件下CO的TOF值分别为22 和25 h-1)显著提高[25].

图9 室温下Au/TiO2催化氧化CO的转化效率
值得说明的是,将未经热处理的Au/TiO2(EG)直接用于CO氧化测试,结果显示材料在暗反应和可见光作用下均没有活性,可能是表面吸附的乙二醇基等有机物种阻碍了CO 分子与界面处晶格氧的反应,而经过热处理后,将材料表面的乙二醇基等吸附物种去除,暴露出CO 的吸附位和晶格氧,使CO 分子能进行吸附活化与氧化.
2.3 CO和O2的化学吸附
2.3.1In situDRIFTS分析 为了得到CO在样品上的化学吸附特征和反应机制,采用了in situDRIFTS进行测试.图10 为TiO2和Au/TiO2样品在暗条件下吸附CO 后的in situDRIFTS 图,从图10(a)可以看到,Au/TiO2样品的红外谱图中位于2 117 和2 173 cm-1的振动谱带为气相中CO 及其在Ti4+上弱吸附的伸缩振动[36-38],CO被快速氧化形成中间物种羧酸盐(),其位于1 670 和1 249 cm-1的振动谱带,进一步快速转化成气相CO2,可在2 360和2 337 cm-1观测到CO2分子不对称伸缩振动谱带[39],也可以较快速度转变成双齿配位碳酸盐(b -)沉积在载体表面[4],其振动谱带位于1 573 cm-1,在表面羟基或少量吸附水存在下,也能快速形成中间物种碳酸氢盐(),可以在1 408和1 223 cm-1观测到相应的振动谱带,而位于1 500 和1 346 cm-1的振动谱带归属为单齿配位碳酸盐(m -)[29,40-41],此外,位于1 643 cm-1的振动谱带归属于载体TiO2表面化学吸附水的弯曲振动谱(δHOH).TiO2载体吸附CO的红外谱图中有气相中CO的伸缩振动,未出现CO2的气相峰,另有一微弱的b -振动谱带,这是CO 与TiO2表面相邻的两个氧原子(O-Ti-O)形成的双齿配位,也表明CO在TiO2表面不会发生氧化反应.

图10 室温下(a)Au/TiO2和(b)TiO2样品吸附CO后的in situ DRIFTS图
2.3.2 TPD分析 采用TPD-MS在线测试可获得CO和CO2的脱附温度,并比较TiO2和Au/TiO2两种样品对CO吸附和转化性能的差异,这里主要监测物种为CO和CO2(与其对应的m/z=28和44).图11是TiO2和Au/TiO2样品在暗条件下吸附CO后TPD过程中的质谱信号曲线图.从图11(a)清楚地看到,在升温过程中出现CO的脱附峰,峰位主要在83°C附近;在CO2的质谱图中,Au/TiO2样品出现明显的CO2的脱附峰(80 ℃),这是CO与TiO2中的晶格氧或表面羟基作用生成类碳酸盐中间物种,在升温过程中脱险成CO2[11-12],与之相比,TiO2样品却未出现类似的脱附峰,再次说明CO不能直接与TiO2中的晶格氧直接转化成CO2.
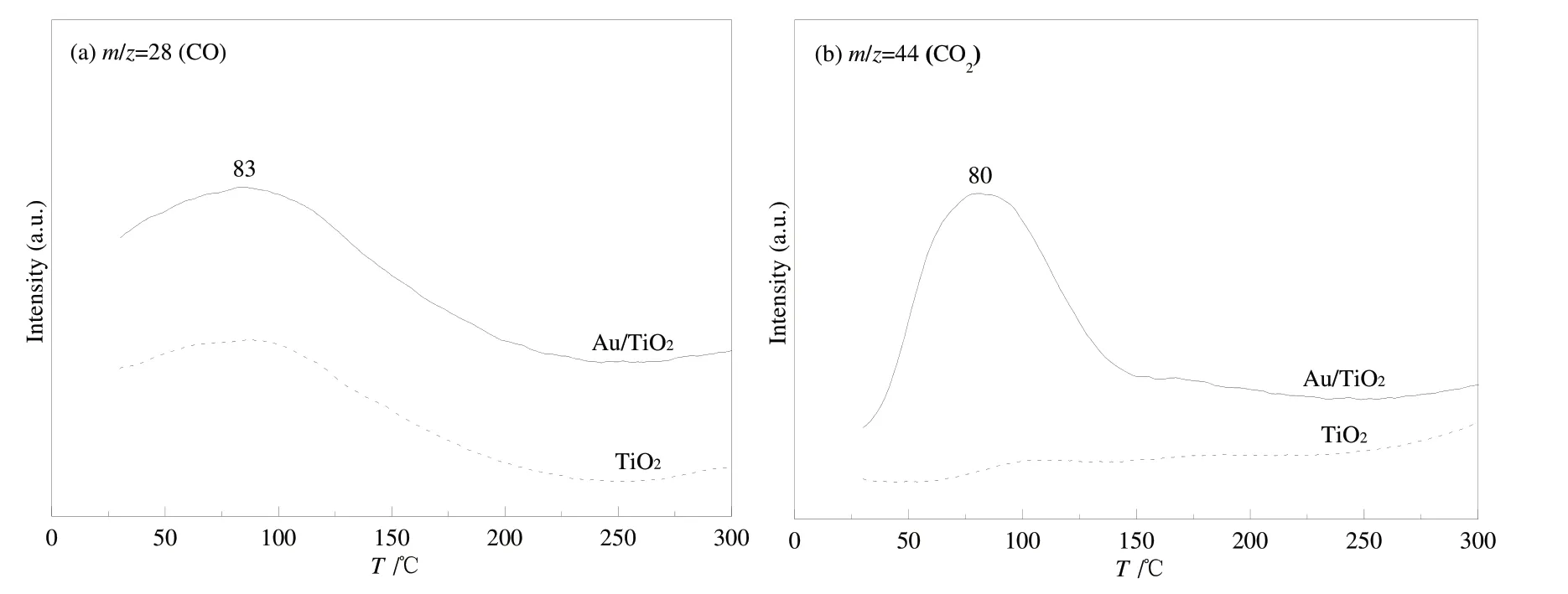
图11 TiO2和Au/TiO2样品吸附CO后TPD过程中的质谱信号曲线
2.3.3 EPR 分析 通过EPR测试可以观测到吸附O2在捕获TiO2导带的电子形成[42],信号强度与数量的变化呈正相关,因此可根据这一变化特征反映出催化剂对氧分子的吸附活化情况.在室温下,Hu[43]等认为氧分子不易在Au位上直接吸附活化和解离成氧原子,他们通过计算得出O2分子在Au粒子(台阶点)上的解离能最低为0.93 eV,也有文献报道在Au-TiO2体界面处的解离能下降到0.52 eV[44].Liu等[45]通过氧同位素交换实验表明Au/TiO2催化剂的表面晶格氧不易与氧分子中的氧原子直接进行交换,而是在CO与晶格氧反应后才起到补充“氧源”作用,这也说明氧分子的吸附活化与CO的吸附转化有直接关系[46-48].
Au/TiO2样品在室温下先进行抽真空预处理,随后立即进行in situESR 测试,结果见图12所示.经过预处理后,在ESR 谱图中观测到不对称的氧空位(Vo)信号峰(g=2.001)[49-51],但没有观测到在Ti4+上的重排峰,而在随后注入CO后,g=2.001的信号增强,说明CO与晶格氧反应后生成更多的氧空位[52,53];同时在g=2.025 出现一逐渐增强的信号峰,这是活化后的在Ti4+上重排[27],而且信号随可见光的引入而进一步增强,说明在可见光的激发下,Au 纳米粒子的LSPR 效应使表面电荷密度增加,促使更多的氧分子生成.结果表明,CO在催化剂表面被氧化后,促进了吸附氧的活化[54,55].图13给出了CO在Au/TiO2上的催化氧化示意图,CO 在Au-TiO2界面与晶格氧化反应最终转化成CO2后,界面处产生新的氧空位,同时氧分子在界面吸附并活化,其中一个氧原子补充晶格氧,另外一个氧原子可能继续补充别的氧空位或迁移到Au粒子表面与活性位上吸附的CO直接反应生成CO2[56-58].

图12 室温下Au/TiO2样品的in situ ESR曲线

图13 可见光作用下CO催化氧化示意图
3 结论
本实验用较缓和的原位光沉积法成功地在钛乙二醇前驱体上负载了Au 纳米粒子,经过热处理后Au 纳米粒子仍能具有粒径较小且不规则形态.在可见光作用下,Au/TiO2催化剂对CO 氧化中表现超高活性,同时具有明显的光促作用和光助稳定性,与传统的沉积沉淀法负载Au相比优势明显.TiO2缺陷位上的电子向Au 纳米粒子表面转移,使Au 电子结合能负移.在可见光作用下,Au 纳米粒子的LSPR 效应使其表面的电子密度进一步增加,对CO 的氧化起到了很好的光促效果.CO 通过消耗Au-TiO2界面处的晶格氧完成氧化过程,同时促进了吸附氧的活化.
