含硅氧烷链段和聚苯醚结构的芳香族聚酰胺的制备及其对环氧树脂的增韧改性
2022-07-14张津怡白小陶
张津怡 白小陶 刘 敏 王 芳 周 权
(华东理工大学 材料科学与工程学院 特种功能高分子材料及相关技术教育部重点实验室,上海 200237)
引 言
环氧树脂(ER)是一类重要的热固性材料,其内部的三维网状结构赋予了它出色的黏接性能、良好的机械性能、优异的化学稳定性和较低的收缩率,因此其广泛用于高性能材料领域,如黏合剂、复合材料的基体和电子封装材料。但是高度交联的网络结构使未经改性的环氧树脂系统变得很脆,导致其对裂纹发生和传播的抵抗力很差,限制了其在高性能材料领域中的应用[1-3]。
改善环氧树脂韧性的方法之一是加入功能化的液体橡胶[4],如羧基封端的丁二烯-丙烯腈橡胶(CTBN)、胺基封端的丁二烯-丙烯腈橡胶(ATBN)和环氧基团封端的丁二烯-丙烯腈橡胶(ETBN)[5-8]。然而,液体橡胶中不饱和结构的存在使得环氧树脂的玻璃化转变温度(Tg)和模量大大降低[9-10]。有研究报道将热塑性塑料添加到环氧树脂中,可以作为橡胶增韧的替代方法[11]。将热塑性塑料如聚醚酰亚胺(PEI)[12]、聚醚醚酮(PEEK)[13]和聚醚砜(PES)[14]添加到环氧树脂中,可以调整整个环氧体系的断裂性能而不降低其玻璃化转变温度[15]。但是在某些情况下,热塑性增韧剂并没有带来环氧断裂韧性的明显改善,甚至由于其与基体的黏附性差而可能导致断裂韧性下降[16]。
聚二甲基硅氧烷(PDMS)是一种很有优势的改性剂,其具有高柔性的Si—O—Si链段、优良的热氧化稳定性、较低的表面张力和良好的耐候性[17]。然而,纯PDMS与环氧树脂前驱体的相容性差,很少单独用作环氧增韧剂[18-19]。利用PDMS与另一环氧可溶链段合成嵌段共聚物可以改善PDMS和环氧树脂的相容性,提升环氧树脂的力学性能。Heng等[20]通过开环聚合和原子转移自由基聚合相结合的方法,利用PDMS合成了一种两亲性多嵌段共聚物,该共聚物在环氧树脂热固性材料中自组装成核壳(core-shell)纳米结构,可以提高环氧树脂的拉伸强度和韧性。Hu等[21]合成了一种新型的聚砜-b-聚二甲基硅氧烷(PSF-b-PDMS)嵌段共聚物,然后将其加入环氧树脂中,通过在环氧基体中形成纳米结构来增韧热固性材料,实验结果表明,PSF-b-PDMS嵌段共聚物改善了环氧热固性树脂的表面疏水性,改性后环氧树脂的韧性与改性前相比几乎提高了3倍。尽管这些研究改善了PDMS与环氧基体的相容性,并且增强了环氧树脂的力学性能,但是同时也导致环氧树脂耐热性能的下降[22-23]。
综合热塑性塑料和PDMS增韧环氧的特点来看,如果能够找到一种环氧相容性好且具备一定耐热性能的热塑性塑料,使其与PDMS反应得到一种增韧剂,就有可能同时发挥热塑性塑料和PDMS的增韧作用,并克服二者各自存在的缺陷。聚苯醚(PPO)是一种工程热塑性塑料,具有低介电常数、低吸水率、优良的热学和力学性能等显著特性,已被广泛应用于环氧网络结构的增韧研究[24-25]。此外,PPO在固化前后均能与环氧基体保持良好的相容性,因此使其与PDMS进行反应,有可能帮助PDMS链段均匀地分散在环氧前驱体中,从而改善两者的相容性。本文以端氨基聚二甲基硅氧烷(ATPDMS)和PPO为原料合成了芳香族聚酰胺(PAPM)增韧剂,并将其引入ER中制备了改性的环氧固化物,研究了PAPM含量对改性环氧固化物的力学性能和热性能的影响,以期为ER在相关领域的应用研究提供参考。
1 实验部分
1.1 实验原料
ATPDMS,阿拉丁试剂有限公司,相对分子质量为600;PPO,相对分子质量为1800,沙伯基础创新塑料有限公司;对苯二甲酰氯(TPC),上海麦克林生化科技有限公司;三乙胺(TEA),上海泰坦科技股份有限公司;双酚A二缩水甘油醚型环氧树脂(E51)、甲基六氢苯酐(MHHPA),上海众何化工技术有限公司;三氯甲烷(CHCl3),国药集团化学试剂有限公司。所用原料的纯度均在99%以上。
1.2 含硅氧烷链段和聚苯醚结构的芳香族聚酰胺的合成
PAPM是由一锅法分两步合成得到的。首先PPO与过量的TPC通过缩聚反应生成由酰氯基团封端的聚酯(PEE),然后PEE与ATPDMS反应得到同时含有硅氧烷链段和聚苯醚结构的PAPM,其合成路线如图1所示。在500 mL四口烧瓶中加入30.0 g PPO(—OH当量为37.5 mmol)和300 mL CHCl3,在常温下机械搅拌30 min使PPO完全溶解,在氮气氛围下加入5.21 mL TEA(37.5 mmol),在冰浴条件下逐滴加入溶有5.71 g TPC(28.13 mmol)的CHCl3溶液,滴加完毕后反应2 h。补加2.61 mL TEA(18.75 mmol),继续滴加溶有2.02 g ATPDMS(—NH2当量为18.75 mmol)的CHCl3溶液,滴加完毕后反应2 h。将反应液倒入1 000 mL分液漏斗中,用去离子水萃取3次,将下层有机相倒入表面皿中,在80℃真空干燥箱中干燥1 h,得到淡黄色固体即为PAPM产物,经凝胶渗透色谱(GPC)测试,其数均相对分子质量为9 169。

图1 PAPM的合成路线Fig.1 Synthesis route of PAPM
1.3 E51/MHHPA/PAPM固化物的制备
E51/MHHPA/PAPM共混物的配方如表1所示。准确称量不同质量比的粉状PAPM产物与E51,加入透明小烧杯中。在150℃油浴中搅拌至粉末溶解,然后降温至80℃,加入一定比例的MHHPA,搅拌均匀以除去气泡。将混合溶液倒入洁净且涂有脱模剂的模具中,按照以下升温程序固化成型:100℃/1 h→130℃/1 h→150℃/1 h→180℃/3 h→220℃/2 h。

表1 E51/MHHPA/PAPM共混物的配方Table 1 Formulation of E51/MHHPA/PAPM blends
1.4 表征与测试
使用Nicolet 5700型傅里叶变换红外光谱仪(FT-IR)(美国Thermo Electron公司)测试样品的红外光谱。测试范围为4 000~400 cm-1,光栅设置为2 cm-1,分辨率为0.09 cm-1。
使用超导傅里叶变换AVANCE500型核磁共振波谱仪(NMR)(德国BRUKER公司)测试样品的NMR氢谱及碳谱。所用溶剂为氘代氯仿(CDCl3),内标为四甲基硅烷(TMS)。
使用Waters1515型凝胶渗透色谱仪(美国Waters公司)测试样品的相对分子质量。所用溶剂为N,N-二甲基甲酰胺(DMF),标样为聚苯乙烯(PS),流速为1.000 mL/min。
使用NETZSCH DSC200F3型差示扫描量热仪(DSC)(德国NETZSCH公司)在高纯氮气保护下测试样品的DSC曲线。温度范围为室温至250℃,升温速率为10℃/min。
使用NETZSCH TG209F1型热失重分析仪(TGA)(德国NETZSCH公司)测试样品的热失重曲线。分别在空气和氮气氛围下测试,温度范围为室温至800℃,升温速率为10℃/min。
使用2 kW CMT 2203型万能拉伸试验机(美国MTS公司)测试样品的弯曲强度。在室温下采用三点弯曲法测试所有固化后的样条,样条尺寸为(120±0.2)mm×(12±0.2)mm×(6±0.2)mm。通过式(1)计算弯曲强度σf,至少测试5个样品,结果取平均值。

式中:Pb为最大破坏载荷,N;L为测试跨距,mm;b为样条宽度,mm;h为样条厚度,mm。
使用2 kW CMT 2203型万能拉伸试验机(美国MTS公司)测试样品的断裂韧性。样品尺寸为60 mm×12 mm×6 mm。使用树脂切割机切出深度为0.25b的v形切口,然后用液氮萃取过的刀片敲击出裂纹,裂纹和缺口的总长度是0.55b至0.65b。利用所测的最大破坏载荷Pb,通过式(2)计算临界应力强度因子KIC,至少测试5个样品,结果取平均值。

式中,f为尺寸系数。
使用DHR-1型动态机械热分析仪(DMA)(美国TA公司)测试样品的储能模量。样品尺寸为60 mm×12 mm×3 mm,温度范围为25~180℃,加热速率为5℃/min,频率为3.0 Hz。
使用S-3400N型扫描电子显微镜(SEM)(日本Hitachi公司)观察固化后样条的表面形貌。样条喷金时间为60 s,测试电压为15 kV。
2 结果与讨论
2.1 PAPM的表征结果
PPO、ATPDMS与PAPM的红外谱图如图2所示。与原料对比可以发现,产物PAPM中1 017 cm-1和1 072 cm-1处的峰归属为ATPDMS中Si—O—Si的伸缩振动,1 268 cm-1处的峰归属于—SiCH3中甲基的变形振动,796 cm-1处的峰归属于Si—C的伸缩振动;1 187 cm-1处的峰归属于PPO中C—O—C的伸缩振动,1 601 cm-1处的峰归属于苯环的骨架振动。此外,产物中羟基峰消失,并在1 737 cm-1处出现了明显的酯羰基吸收峰,3 316 cm-1处的峰为酰胺基团上N—H的伸缩振动峰,1 655 cm-1和1 544 cm-1处的峰分别为酰胺Ⅰ带和酰胺Ⅱ带吸收峰,表明TPC上的酰氯基团成功地与PPO中的羟基和ATPDMS上的伯胺反应,成功合成了产物PAPM。
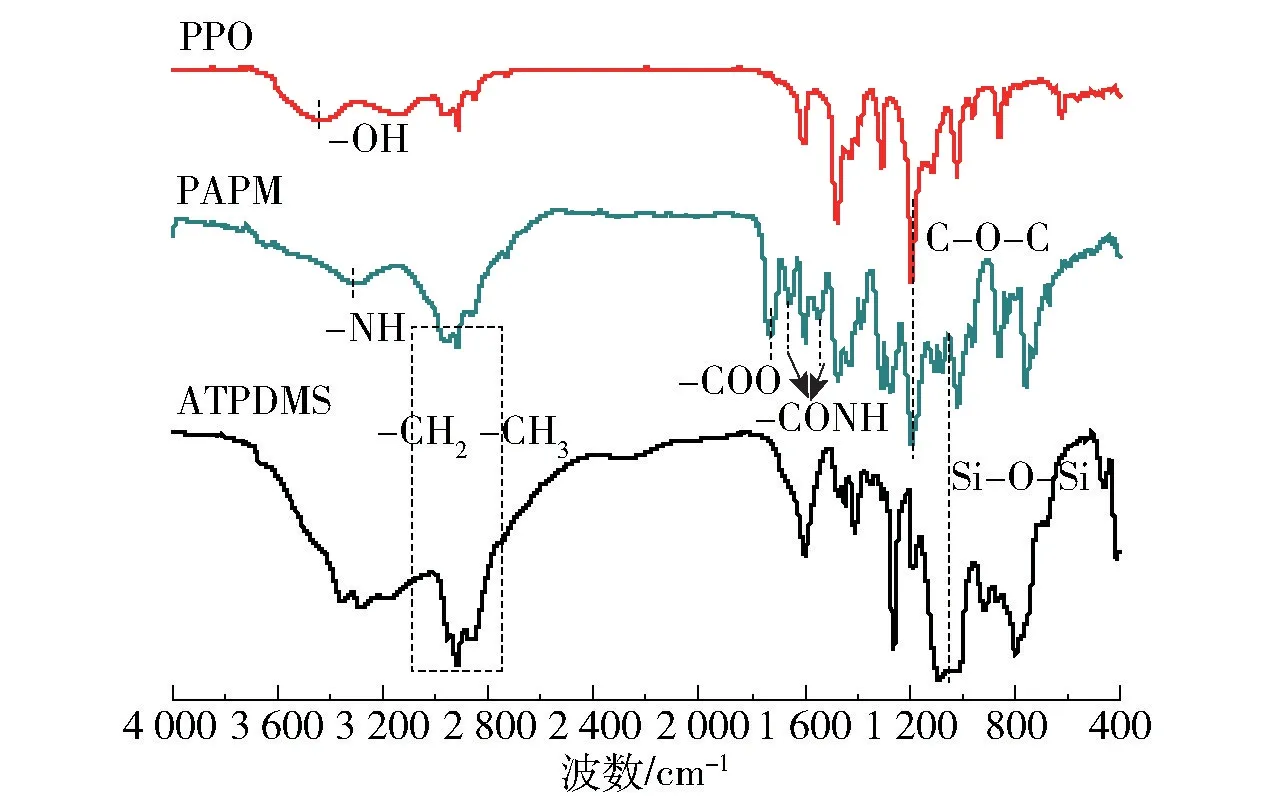
图2 PPO、ATPDMS与PAPM的FT-IR谱图Fig.2 FT-IR spectra of PPO,ATPDMSand PAPM
图3是PAPM的13C NMR谱图。75.97处的信号峰是CDCl3的化学位移。ATPDMS上甲基与亚甲基的化学位移分别为:(a)0.09,(b)7.54,(c)60.57,(d)13.20,(e)29.92和(f,g)44.43。PEE中不同苯环上碳原子的化学位移分别位于(i)~(y)(113.31~153.56)。值得注意的是,(l)162.91处出现了酯基碳原子的信号峰,(h)164.92处出现了酰胺基团碳原子的信号峰,说明PPO两端的羟基成功地与TPC反应,生成了含有酯基的PEE,而PEE又进一步与ATPDMS反应,生成了聚酰胺。

图3 PAPM的13 C NMR谱图Fig.3 13 CNMR spectrum of PAPM
图4是PAPM的1H NMR谱图。7.19处出现的峰是CDCl3的化学位移峰,(a)0.01是ATPDMS中与Si相连的甲基上的质子峰,(b)1.18、(c)3.07、(d)1.32、(e)2.91、(f)2.99和(g)4.33分别是ATPDMS主链上不同亚甲基的质子峰,(i)8.03、(j)8.19、(l)6.40、(q,r)6.91是PEE苯环上的质子峰。可以看出,PAPM产物中不仅包含几种原料的质子峰,而且还在(h)8.30处出现了酰胺基团上仲胺的质子峰,进一步说明了PEE两端的酰氯基团成功地与ATPDMS反应,生成了产物聚酰胺。

图4 PAPM的1 H NMR谱图Fig.4 1 H NMR spectrum of PAPM
2.2 PAPM对E51的共固化作用
图5是不同PAPM含量的E51/MHHPA/PAPM体系在固化前的DSC曲线。不含PAPM的E51/MHHPA体系的最大固化放热温度(Tmax)为178.7℃,含有5%、10%、15%PAPM的环氧混合体系的Tmax分别为183.4℃、179.2℃、173.4℃,与不含PAPM的体系相比变化不大。不含PAPM时,E51/MHHPA体系的放热量仅为60.5 J/g;加入PAPM后,E51/MHHPA/PAPM体系的放热量增大,且固化峰变宽。含有5%、10%、15%PAPM的环氧混合体系的放热量分别为88.69、179.6、203.9 J/g,放热量的明显变化说明PAPM的加入可能使得整个环氧体系的固化机理发生了改变,从而起到共固化的作用。不含PAPM时,E51树脂仅由MHHPA固化,E51中的羟基引发MHHPA开环生成羧酸,其与环氧基反应生成羟基,羟基继续与酸酐反应,进而一步步地形成交联的网状结构[26]。加入PAPM改性后,PAPM中的酰胺基团首先与E51发生开环反应生成叔胺,此时叔胺可以起到环氧促进剂的作用,因此其在环氧网络中的反应遵循叔胺类促进剂的固化机理[27],即叔胺进攻MHHPA形成羧酸盐阴离子对,该阴离子对与环氧基进行加成,生成一个新的含有酯基的羧酸盐离子对,这个羧酸盐离子对又可以与MHHPA生成一个新的离子对,然后按照阴离子机理继续交替反应而逐步固化。
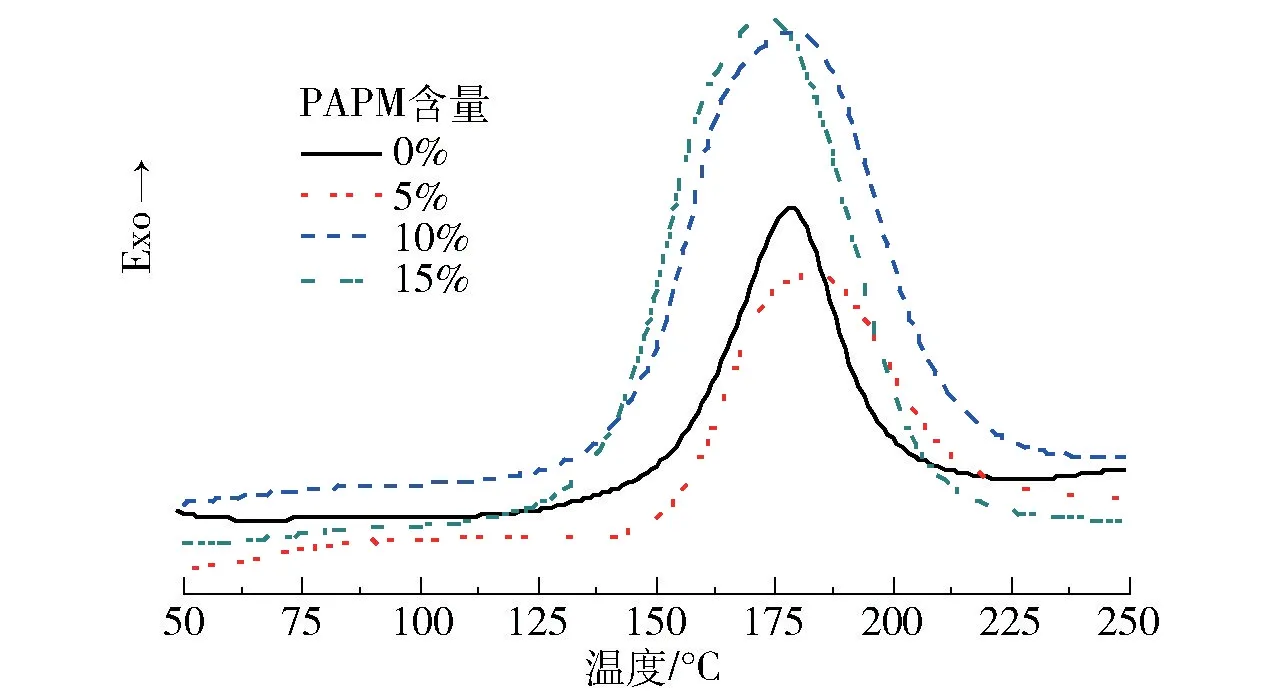
图5 不同PAPM含量的E51/MHHPA/PAPM体系在固化前的DSC曲线Fig.5 DSC curves of E51/MHHPA/PAPM systems with different PAPM contents before curing
2.3 PAPM与E51的相容性
在E51中分别添加质量分数为5%的ATPDMS、PEE和PAPM,测试其相容性,结果如图6所示。由图6(a)可见,固化前ATPDMS在E51中呈现出白色浑浊的不透明状态,表明二者不相容。图6(b)中,固化前PEE与E51共混时呈现出均一透明的状态,表明二者的相容性良好。由图6(c)可见,PAPM可以溶解于E51中,其混合溶液呈淡黄色,介于E51/ATPDMS与E51/PEE之间,说明E51/PAPM体系在可见光尺度上没有发生相分离,这是由于环氧可溶链段PEE对环氧不溶链段PDMS的拉扯作用使得PAPM可以很好地溶解在E51中。图7为不同PAPM含量的E51/MHHPA/PAPM固化物照片。不含PAPM的E51/MHHPA固化物在固化后是透明的。随着PAPM含量的增加,环氧固化物的颜色逐渐加深,当PAPM含量为15%时,环氧固化物始终保持透明的状态,说明PAPM与E51在固化后仍然是相容的,没有发生宏观可见光尺度上的相分离。

图6 ATPDMS、PEE、PAPM与E51在固化前的相容性Fig.6 Compatibility of ATPDMS,PEE,PAPM and E51 before curing

图7 不同PAPM含量的E51/MHHPA/PAPM固化物照片Fig.7 Photos of E51/MHHPA/PAPM thermosets with different PAPM contents
2.4 E51/MHHPA/PAPM固化物的力学性能
图8是不同PAPM含量的E51/MHHPA/PAPM固化物的临界应力强度因子KIC。从图中可以看出,不含PAPM的E51/MHHPA体系固化后的KIC值仅为0.90 MPa·m1/2,随着PAPM含量从5%增加到15%,环氧固化物的KIC值由1.44 MPa·m1/2增加到1.91 MPa·m1/2,PAPM含量为15%的环氧固化物的KIC比不含PAPM的环氧体系提高了112.2%。这是因为PAPM同时含有聚苯醚结构和聚二甲基硅氧烷链段,聚苯醚作为一种热塑性塑料,其本身就具有增韧环氧树脂的作用,而聚二甲基硅氧烷也是一种有效的环氧增韧剂,二者的协同增韧作用使得E51/MHHPA/PAPM固化物的断裂韧性得到大幅度的提高。

图8 不同PAPM含量的E51/MHHPA/PAPM固化物的KICFig.8 KIC of E51/MHHPA/PAPM thermosets with different PAPM contents
图9是不同PAPM含量的E51/MHHPA/PAPM固化物的弯曲强度。由图可知,未加PAPM时,环氧树脂固化物的弯曲强度为114.6 MPa;加入PAPM后,弯曲强度变化的幅度不大。当PAPM含量为5%时,环氧固化物的弯曲强度降到最低值(106.6 MPa),这可能是由于PAPM的引入在环氧基体内部形成应力集中点,导致材料在受到外力时其内部应力分布不均匀,从而引起材料的脆性断裂。当PAPM含量高于5%时,环氧固化物的弯曲强度略有提高,可能是因为PAPM中的仲胺基团与环氧基体反应,使得PAPM与基体缠结得更加紧密,集中的应力被部分分散到环氧基体内部,从而稀释了PAPM作为应力集中点产生的应力,使得环氧固化物的弯曲强度的下降趋势有所回升。
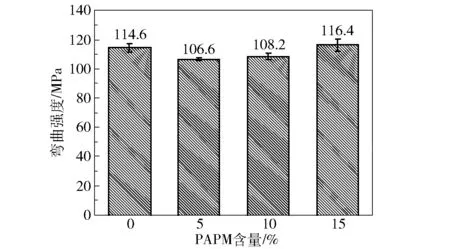
图9 不同PAPM含量的E51/MHHPA/PAPM固化物的弯曲强度Fig.9 Bending strengths of E51/MHHPA/PAPM thermosets with different PAPM contents
为了进一步探究PAPM对环氧树脂的增韧机理,本文对环氧固化物弯曲断面的形貌进行了表征。图10是不同PAPM含量的E51/MHHPA/PAPM固化物弯曲样条断裂面的SEM图。从图10(a)中可以看出,在未经改性的环氧树脂的断裂面表面分布着长而深的直条状裂纹,经局部放大可见相邻裂纹之间的沟壑较深,裂纹走势基本一致,且彼此之间互不影响,表明受到外力时,不添加PAPM的环氧树脂的表面裂纹发展迅速,几乎横贯整个断裂面,并且有向环氧树脂内部发展的趋势,整体表现为脆性断裂。如图10(b)所示,当PAPM含量为5%时,环氧固化物的裂纹浅浅地分布在其断裂面表面,其中一些长裂纹不再笔直,在发展过程中出现明显的波纹状转折,同时伴随着从主裂纹上分叉产生的微裂纹,而另一些裂纹发展到中途直接停止,说明PAPM的加入在环氧基体内部引入了应力集中点,可以有效地吸收外部能量,改变甚至阻止裂纹的发展,从而对环氧树脂起到增韧的作用。由图10(c)可以看出,当PAPM含量增加到10%时,条形的主裂纹逐渐封闭,转变为环形状态,此时的微裂纹细而短小,并且绝大多数被环形的主裂纹包围在内部,断裂面整体呈现出一种“漩涡”结构。如图10(d)所示,当PAPM含量为15%时,改性后的环氧树脂断裂面的漩涡状裂纹结构更加丰富,几乎所有的微裂纹都被包裹在其中,这种漩涡状裂纹在外力作用下可以有效地吸收能量,从而进一步阻止微裂纹向外部扩散。后两种情况下环氧固化物的断裂均为韧性断裂,尤其在15%PAPM含量下环氧固化物的断裂面普遍可见颗粒状的环氧树脂碎屑,说明此时经改性的环氧树脂已具有相当的韧性。PAPM的仲胺与环氧基团反应所生成的叔胺结构在一定程度上起到锚钉的作用,可以将部分外力过渡给环氧基体,使得改性材料整体对应力起到均匀分散的作用,因此需要施加更大的外力才能使其断裂。
图11是不同PAPM含量的E51/MHHPA/PAPM固化物的储能模量曲线。未添加PAPM时,25℃下E51/MHHPA固化后的储能模量为2 299.2 MPa;随着PAPM添加量的增加,环氧固化物的储能模量先增大后减小;当PAPM添加量为5%时,25℃下储能模量达到最大值(3 600.9 MPa),相比不含PAPM的E51/MHHPA体系提高了56.6%。这是因为当环氧树脂中PAPM含量较低时,PEE链段中的刚性苯环结构对材料力学性能的影响起主要作用,因此改性材料的储能模量得到提高。当PAPM含量增加到10%和15%时,25℃下环氧固化物的储能模量分别降至3 297.3 MPa和3 228.9 MPa。这是因为PAPM含量增加后,体系中的柔性链段如醚键、酯基和酰胺键的比例提升,导致改性材料的储能模量下降,但仍高于不含PAPM的E51/MHHPA体系,说明芳香族聚酰胺的加入对提高环氧树脂的刚性有一定的作用。
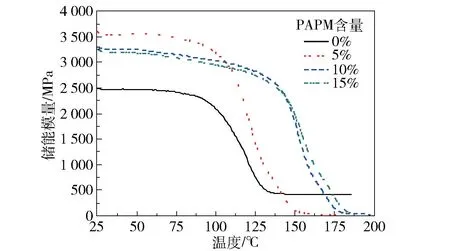
图11 不同PAPM含量的E51/MHHPA/PAPM固化物的储能模量曲线Fig.11 DMA curves of storage modulus of E51/MHHPA/PAPM thermosets with different PAPM contents
2.5 E51/MHHPA/PAPM固化物的热性能
图12是不同PAPM含量的E51/MHHPA/PAPM固化物在空气氛围和氮气氛围下的TGA曲线。从图12(a)中可以看出,在空气氛围下,环氧固化物的热降解过程分为两个阶段:300~450℃阶段发生的降解反应主要是环氧树脂主链的碳化和侧链的氧化,450~650℃阶段发生的降解反应是芳环的氧化或碳化。PAPM含量为0%、5%、10%、15%的环氧固化物失重5%的温度(Td5)分别为332.8、352.9、353.1、355.9℃,表明改性后的环氧固化物的Td5高于改性之前,这是因为PAPM中的苯环结构在第一个降解阶段起着耐高温的作用。从图12(b)中可以看出,在氮气氛围下,PAPM含量为0%、5%、10%、15%的环氧固化体系在800℃时的质量残留率分别为3.95%、7.50%、7.72%、7.94%。添加PAPM的环氧固化物的质量残留率高于未改性的环氧固化物,这是因为PAPM中存在大量的Si—O键,Si—O键的键能较高,能够赋予环氧固化物一定的耐热氧化性能。此外,有机硅在高温区可以生成一层稳定的保护膜,覆盖在环氧树脂表面,使得其残余质量率增加。

图12 不同PAPM含量的E51/MHHPA/PAPM固化物在空气和氮气氛围下的TGA曲线Fig.12 TGA curves of E51/MHHPA/PAPM thermosets with different PAPM contents in air and nitrogen atmospheres
图13是不同PAPM含量的E51/MHHPA/PAPM固化物的DSC曲线。可以看出:添加PAPM的环氧固化物的Tg高于未改性的环氧固化物;当PAPM含量为5%时,环氧体系的Tg的提升速度最大,随后逐渐变缓;当PAPM含量为15%时,Tg达到最大值(126.7℃),比不含PAPM的环氧固化物提高了28.2℃。PAPM的加入大幅度提高E51树脂的Tg的原因主要有:1)虽然PAPM中含有低Tg的硅氧烷链段,以及反应时生成的柔性酯基和酰胺基团,但是PPO中含有耐热的苯环结构,对环氧树脂Tg的提高可能抵消甚至大于柔性链段和基团对环氧树脂Tg的降低作用;2)PAPM与环氧基团的开环反应使得环氧固化体系的交联密度提高,从而形成了更加致密的三维网络结构。因此,加入5%PAPM的环氧固化物的Tg提升幅度较大。当PAPM含量超过5%时,Tg的这一升高趋势有所变缓,这是因为随着固化反应进行到一定程度,分子链变得僵硬,环氧分子很难穿过层层网络与羧基阴离子接触,只能作为大分子被包裹在网络中,体系的交联密度难以进一步提高。
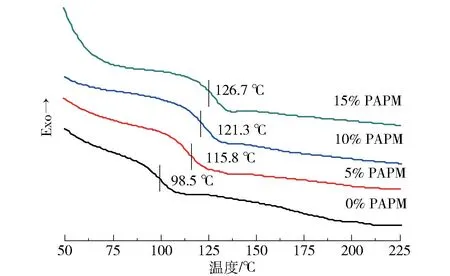
图13 不同PAPM含量的E51/MHHPA/PAPM固化物的DSC曲线Fig.13 DSCcurves of E51/MHHPA/PAPM thermosets with different PAPM contents
3 结论
采用一锅缩聚法成功制备了一种含硅氧烷链段和聚苯醚结构的芳香族聚酰胺PAPM,使得具有增韧效果和耐高温性能的链段共存于大分子中,并将其作为改性剂,通过仲胺与E51的基团反应以及MHHPA的固化作用固定在环氧基体中,改善了E51的力学性能和耐热性能。测试结果表明,当PAPM添加量为15%时,环氧固化物的KIC相比不添加PAPM的环氧体系增加了112.2%;当PAPM添加量为5%时,环氧固化物的储能模量相比不添加PAPM的环氧体系增加了56.6%;当PAPM添加量为15%时,环氧固化物的Tg相对于不含PAPM的环氧体系提高了28.2℃。
