一例鲁宾斯坦-泰比综合征患者的临床及基因分析并文献复习
2022-07-05万孝苗黎玲刘昌伟申超辉吴利雯周素娴
万孝苗,黎玲,刘昌伟,申超辉,吴利雯,周素娴*
鲁宾斯坦 - 泰比综合征(Rubinstein-Taybi syndrome,RsTs)是于1963年首次被儿科医生RUBINsTEIN等[1]报道的一种罕见的多系统发育障碍疾病,患者临床表现为不同程度的智力低下、生长发育迟缓、小头畸形、拇指宽大和第一脚趾宽大及独特的面部特征(包括额部发际线低、弓形眉毛/粗眉、下倾的睑裂、突出的喙鼻、鼻下小柱、耳朵低位、上唇薄、高腭弓、小颌、牙齿畸形、尖牙、“鬼脸”微笑等)。RsTs患者常伴有累及多系统多器官的多种先天性畸形(胼胝体发育不全、小脑蛭部畸形、弱视、泪道异常、先天性心脏病)、内分泌系统障碍(甲状腺/垂体发育不良)、胃肠疾病(胃食管反流/巨结肠)、肾脏畸形和隐睾等,甚至会累及皮肤(如毛母细胞瘤、瘢痕疙瘩形成倾向、多毛症)[2]。有研究发现RsTs可累及神经组织、间叶组织和淋巴造血组织,导致患者患癌倾向增加(如神经母细胞瘤、横纹肌肉瘤、淋巴瘤和白血病),且发病率远低于一般罕见病(<1/100 000)[3],智力低下人群RsTs患病率较智力正常人群高[4]。本文通过对2019年桂林医学院附属医院收治的1例疑似RsTs的患者及其双亲进行全外显子组基因测序,旨在通过描述该患者临床特征及基因测序结果,提高临床医生对该病的认知,减少误诊和漏诊,为该病基因型-表型相关性研究、RsTs的诊治提供更多参考。本研究通过桂林医学院附属医院伦理委员会许可(伦审号:YJsLL202131),患者父母均知情同意。
1 病例简介
患者,女,16岁,因“发现生长发育异常10余年”于2019年10月至桂林医学院附属医院内分泌科就诊。患者系足月顺产,母亲妊娠期间有服用排结石药物史,具体药物及使用剂量不详,妊娠过程相对顺利。患者父母仅记其出生体质量为3.0 kg,其余出生情况记不清,父母自觉患者出生时无明显异常。患者出生后经母乳喂养至2岁,期间按时添加辅食。患者2岁时开始走路,3岁时开始缓慢说话,直至5~6岁才能讲述完整句子,该情况未引起患者父母重视,未察觉患者发育异常。8岁时患者父母发觉患者身高相比同龄人偏矮小,未行诊治。目前患者智力低下,语言表达能力差,学习不佳,运动尚可,反应较同龄人差。患者10余年前曾行“右眼泪管探通术”,近5年来数次出现皮肤瘙痒,有时累及嘴唇,出现红肿,始终未能明确病因。其余无特殊。患者13岁时月经初潮,不规律,每次持续3~4 d,具体经量、颜色不详。患者父母身体康健,否认家族遗传病史。
体格检查:生命体征平稳,体质量38 kg,身高132 cm,上/下部量分别为68 cm、64 cm。智商低,反应迟钝,仅能计算个位数加减乘除法,吐词尚清楚。有特殊面容,头发浓密,粗眉,眼距增宽,内眦赘皮,突出的喙鼻,鼻下小柱,上唇薄,高腭弓,颈部粗短,后发际低平,后颈部毳毛多,大拇指和第一脚趾宽大(图1)。四肢毛发多。心肺腹查体无明显异常。
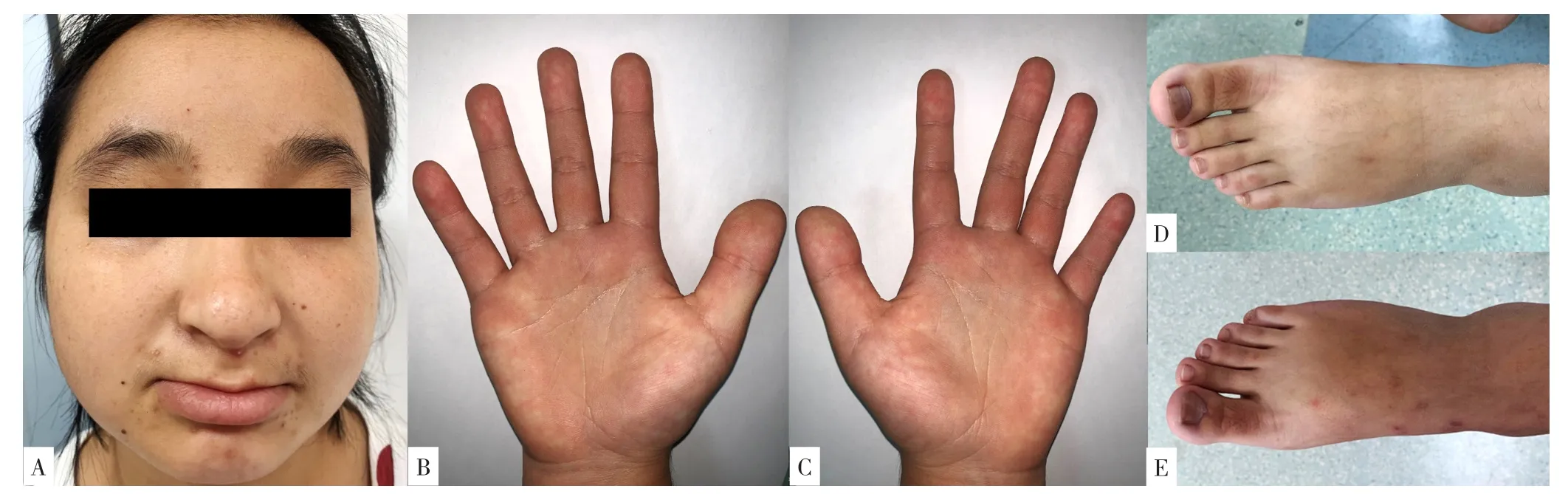
图1 患者面容及手足特征Figure 1 Facial,hands and feet features of a 16-year-old female with Rubinstein-Taybi syndrome
实验室检查:血尿常规、大便常规、生化、皮质醇、胰岛素、醛固酮、甲状腺/甲状旁腺/性腺/生长激素等未见异常。心腹超声、垂体MRI平扫、胸片无异常。右侧髋关节骨密度为0.850 g/cm2,Z 值为-0.7 sD。跟骨侧位、手掌正侧位 DR:左足、手掌骨质均未见异常,骨骺线均已完全闭合。
为进一步明确诊断,经患者监护人知情同意,取患者及其父母外周静脉血各3 ml,由深圳华大临床检验中心进行全外显子捕获测序(WEs)检测。检测方式:以受检者的血液DNA样本为检测材料,首先将样本DNA打断,然后制备文库,通过 BGI V4 芯片捕获和富集目标基因的外显子及其相邻剪切区的DNA,最后通过 MGIsEQ-2000 测序平台进行基因突变检测。检测结果:测得原始数据约21.1G,目标区长度约58.68 Mb、覆盖率99.72%,目标区平均深度187.08 X,平均深度>20X 位点达98.27%;16号染色体上CREBBP基因突变:c.3832G>A(p.Glu1278Lys) 。依据 ACMG 指南,该变异被判断为致病变异。目前已有该突变致病性的相关报道[5]。其父母未检测出该基因突变,考虑患者为新发突变。全外显子基因测序结果见图2。
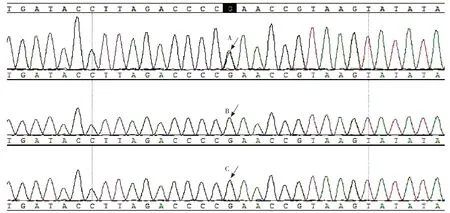
图2 全外显子基因测序结果Figure 2 Whole exome sequencing results of a 16-year-old female with Rubinstein-Taybi syndrome and her parents
2 文献检索策略及结果
以“鲁宾斯坦-泰比综合征/Rubinstein-Taybi 综合征/RsTs综合征”“鲁宾斯坦-泰比综合征/Rubinstein-Taybi综合征/RsTs综合征 and CREBBP基因”“鲁宾斯坦-泰比综合征 /Rubinstein-Taybi 综合征 /RsTs 综合征 and EP300 基因”为关键词在中国知网、万方数据知识服务平台、维普网中检索2001—2021年发表的中文文献。同时以“Rubinstein-Taybi syndrome/RsTs”“Rubinstein-Taybi syndrome/RsTs and CREBBP”“Rubinstein-Taybi syndrome/RsTs and EP300”为关键词,在PubMed 中检索2001年1月至2021年11月发表的英文文献。纳入标准:(1)对多病例进行详细分析的公开发表论文;(2)在动物、人体细胞进行有效研究的公开发表论文;(3)全面、详细的综述报道。排除标准:(1)单一的病例报道;(2)研究结果相似的论文。最终检出符合要求的英文文献29篇。
3 讨论
RsTs是一种非常罕见的常染色体显性遗传病,临床表现多样,常见表现有智力障碍、发育迟缓、小头畸形、拇指和第一脚趾宽大及特征性面容等,可累及多器官、多系统。
目前研究发现,RsTs主要是因CREBBP(OMIM#600140)和EP300(OMIM#602700)这两个普遍表达且高度保守的同源基因发生突变所致。多项研究发现,55%~65%的RsTs由CREBBP基因突变引起[6-8],其中约80%与致病序列变异有关,约 20%与缺失片段多少有关[2]。NEGRI等[9-11]发现5%~10%的RsTs是因EP300基因突变所致,其中约50%为移码突变,缺失型较少见[2]。在CREBBP和EP300基因突变患者中出现频率最高的是点突变(即移码、无义、错义和剪接位点),其次是缺失(包括基因内、全基因或扩展到邻近区域)、易位和倒位[12-13]。综上所述,仍有部分RsTs患者的发病机制尚不清楚,该综合征的病因和发病机制需进一步研究。
CREBBP基因位于染色体16p13.3上,有31个外显子,大小约为150 kb,其可编码一个分子量为2 6531Da、由2 442个氨基酸组成的多功能转录辅助激活蛋白CREB结合蛋白(CBP,又名cAMP反应元件结合蛋白),其突变所致RsTs被定义为 RsTs 1型(RsTs1;OMIM#180849)。EP300基因位于染色体22q13.2上,外显子数目同CREBBP基因,大小约为88 kb,编码p300蛋白,其突变所致RsTs被定义为RsTs 2 型(OMIM#613684)。FERGELOT 等[14]研究指出,RsTs 2型患者除了认知功能水平相对更高及发生先兆子痫风险更高外,其余临床表现均较RsTs 1型患者略轻。两基因皆可编码组蛋白乙酰转移酶(HAT)。CREBBP和EP300基因编码的CBP和p300蛋白是大量DNA结合转录因子的转录共激活因子,已在16 103个人类基因的启动子区域被证实[15]。在这两个共激活因子中有一个非常重要的HAT结构域[2],与RsTs的发生有很大关联。据报道,CBP和p300在整个蛋白质水平上相似性为57%,但在HAT结构域的相似性却达88%[16],二者可与400多个蛋白质相互作用,可乙酰化转录因子,如P53、核酸内切酶APE1、DNA聚合酶、DNA糖基化酶TDG等,参与调节细胞生长、分化、DNA修复、凋亡、神经元的可塑性和肿瘤抑制[17]。此外,CBP和p300还可利用HAT乙酰化组蛋白H3和H4 N端赖氨酸,中和其侧链正电荷,解开DNA和组蛋白相互作用,促进DNA转录[18-19]。
KORZUs等[20]首次证明CBP的HAT活性是脑信息处理所必需的,同时认知功能的特定过程受去乙酰酶/乙酰化酶活性对组蛋白的调节的影响。ALARI等[21]从3例CREBBP基因和2例EP300基因突变患者的外周血细胞中建立了RsTs的体外诱导多功能干细胞(IPsC)神经元模型,观察到患者的神经元形态学改变和神经元兴奋性降低可能是导致RsTs患者认知障碍的潜在病理机制之一。CALZARI等[22]也利用RsTs1和RsTs2患者外周血中的IPsC来源的神经元模拟RsTs,通过与健康对照组进行差异表达基因(这些基因标志着IPsC来源的神经前体细胞向皮质神经元转变)比较,结果发现RsTs患者细胞中的神经前体细胞的神经元转录程序存在缺陷和改变,并指出这些转变可能与RsTs患者的认知障碍有关。ALARI等[23]通过研究RsTs患者IPsC衍生的神经元模型发现,组蛋白去乙酰化酶抑制剂(HDACi)可改善RsTs患者IPsC神经元的形态缺陷和低兴奋性。在体外RsTs患者的淋巴母细胞系中,丁酸钠(NAB,一种具有很强HDACi活性的天然短链脂肪酸)可以改善CBP/p300相关的乙酰化缺陷,并可部分挽救果蝇CBP突变体的RsTs表型[24]。BABU等[25]建立了斑马鱼RsTs模型,在使用HDACi Ⅲ(一种肿瘤特异性抗原的酰胺类似物)和CHIC35(一种赖氨酸去乙酰化酶的抑制剂)后发现HAT结构域的过度表达可以挽救斑马鱼RsTs模型中的部分表型,如颅面软骨和胸鳍缺陷,表明通过激活HAT或抑制HDAC可能是治疗乙酰基转移酶缺陷相关疾病的有效方法。
研究发现,点突变主要影响CBP的HAT结构域,即HAT 活性的降低足可引发 RsTs[5]。CROss 等[26]研究指出,CBP HAT结构域中的错义突变是致病性的。经基因检测发现,本例患者16号染色体上的CREBBP基因HAT区发生错义突变,c.3832G>A(p.Glu1278Lys),据此并结合患者的临床表现,可明确诊断为RsTs。鉴于患者父母没有发生基因突变,且家族中没有类似情况,表明患者为新发突变。CREBBP基因在尚在发育的皮肤、心肺、肝脏以及血管系统等的特定细胞类型中均有表达,当CREBBP基因发生突变时,其所编码的CBP和HAT不能乙酰化组蛋白,以致DNA转录失控,表达出不适当的蛋白质。患者近5年来出现不明原因的皮肤瘙痒,不排除与RsTs有关。
CREBBP基因突变大多数为新发突变,其复发率较低。尽管该病表现为常染色体显性遗传,但由于RsTs患者结婚生育后代的概率降低,RsTs家庭聚集现象较为少见。RsTs对男性和女性的影响基本相同,产前检查多可见胎儿发育正常,出生时各项检查指标可达到正常新生儿平均值或者接近正常新生儿,之后患儿在婴儿期开始出现生长发育迟缓,青春期可出现肥胖或超重倾向[3]。关于成年RsTs患者寿命的报道不多,一般而言大部分患者的预期寿命接近正常人,而若患者伴严重先天性心脏病或因免疫低下导致呼吸道感染等症状反复发生,则会直接影响其寿命。多项研究表明,RsTs患者的基因型与表型没有明确关联[27-28],但仍有研究者继续致力于基因型与表型的相关性研究。
临床提示:
鲁宾斯坦-泰比综合征(Rubinstein-Taybi syndrome,RsTs)十分罕见,临床表现多样,并可累积多器官、多系统。目前尚无针对性指南,部分患者可通过临床特征和基因检测确诊,但仍有不少患者无法确诊。RsTs易误诊为特雷彻·柯林斯(Treacher-Collins)综合征、哈勒曼-斯特雷夫(Hallermann-streiff)综合征、Pfeiffer综合征、胎儿面容综合征(Robinow syndrome)等,常导致该病不能被及早诊断、及时治疗。本例患者起病隐匿,加上家长疏忽,未能及早诊治,错过了最佳治疗时机。
目前除对症治疗外尚无治疗RsTs的更好方式,因此临床仍需进一步研究相关治疗手段。在RsTs患者的诊疗及预防方面,首先可通过定期产检及产前遗传学筛查等方式及时发现疑似RsTs胎儿;其次可在患儿出生后定期进行听力、眼、牙齿、骨骼、神经精神、心脏、肾脏、脑等方面的医疗评估,指导患儿的医疗护理及康复训练。对于成年RsTs患者,目前并没有标准的医疗管理指南,但除应关注患者的常规身体健康状况外,也要注意患者的神经精神心理变化(如焦虑、自闭、强迫症、情绪不稳定等)。
国内外有关RsTs患者的报道虽已超1 000例,但实际临床对该疾病的相关研究依然较少,也尚无对症治疗的特效药物。目前仅在细胞和动物水平上发现使用组蛋白去乙酰化酶抑制剂(HDACi)对CREBBP和EP300基因HAT结构域突变所致RsTs有逆转作用,但尚无法直接应用于临床治疗[24-25]。有一部分RsTs患者基因诊断结果不明确,不能排除可能存在尚未发现的其他发病机制。未来应在细胞、动物等领域方面加强相关研究,以期找到合适的药物或方法治疗RsTs及其他罕见病患者,减少他们的疾病痛苦。
作者贡献:万孝苗负责文章构思与设计,撰写及修订论文,对文章整体负责;万孝苗、黎玲进行资料、文献的收集与整理;刘昌伟、申超辉、吴利雯参与论文的修订;周素娴负责文章的质量控制及审校。
本文无利益冲突。
