Pediatric acute viral hepatitis with atypical variants:Clinical dilemmas and natural history
2022-07-04MoinakSenSarmaAathiraRavindranath
Moinak Sen Sarma,Aathira Ravindranath
Moinak Sen Sarma,Department of Pediatric Gastroenterology,Sanjay Gandhi Postgraduate Institute of Medical Sciences,Lucknow 226014,Uttar Pradesh,India
Aathira Ravindranath,Division of Pediatric Gastroenterology,Apollo BGS Hospitals,Mysuru 570023,Karnataka,India
Abstract Classical acute viral hepatitis(AVH)has an uncomplicated outcome.Acute liver failure has a grave prognosis.Atypical manifestations of AVH are a group of disorders that causes significant morbidity and dilemmas in children.These include prolonged cholestasis,relapsing hepatitis,ascitic form of AVH,late-onset hepatic failure(LOHF),intravascular hemolysis,and provoking an autoimmune trigger leading to autoimmune hepatitis.These entities cause significant liver dysfunction or worsening and are often difficult to differentiate from chronic liver disease(CLD).Ascitic form of AVH,LOHF,decompensated CLD and acute-onchronic liver failure have significant overlapping features that need to be carefully dissected out.In many cases,only on long-term follow-up,these clinical entities can be separately identified.Intravascular hemolysis is usually caused by associated glucose-6-phosphate dehydrogenase deficiency.Rarely CLD such as Wilson disease and autoimmune hepatitis can also present with hemolysis in the initial presentation,which can mimic AVH with hemolysis.Identifying deviations from typical manifestations aid in avoiding unnecessary investigations,allowing focused therapy and alleviating anxiety.
Key Words: Viral;Hepatitis;Atypical;Cholestasis;Relapsing;Hemolysis;Ascites
lNTRODUCTlON
Ancient Sumerians considered the liver to be the seat of the soul and jaundice to be caused by the devil.From that era,we have come a long way in understanding hepatitis.Although the transmissible nature of hepatitis was known even in the Middle Ages,discovery of the Australia antigen and subsequently the other hepatotropic viruses formed the watershed in the history of acute viral hepatitis(AVH)[1].While the majority of AVH has a benign self-limiting course with spontaneous resolution,acute liver failure(ALF)has a fulminant pattern with poor recovery of native liver.In between the two extremes of these phenotypes lies a major subgroup of complications that are collectively known as atypical manifestations of AVH(Figure 1).These entities have a variable outcome.They include complications such as prolonged cholestatic phase,relapsing hepatitis,ascitic variant of AVH,hematological involvement(immune thrombocytopenia,intravascular hemolysis,and aplastic anemia)and extrahepatic multisystemic issues(acute kidney injury,pancreatitis,Guillain-Barré syndrome,myocarditis,and myositis)[2].In addition late-onset hepatic failure(LOHF),a debated entity closely mimics acute-onchronic liver failure(ACLF).Due to the overlapping presentation,it is a challenging task to distinguish AVH from underlying chronic liver disease(CLD)(Figure 2).Of all the atypical features,this review is limited to the discussion of those uncommon features of AVH that are associated with liver dysfunction or worsening.
EPlDEMlOLOGY
The prevalence of AVH is variable but atypical manifestations are encountered in 14%-22% of children[3].Etiology-specific prevalence is 30% in hepatitis A virus(HAV),15% in hepatitis E virus(HEV)and 3% in hepatitis B virus(HBV)infections[3].The wide range of prevalence is possibly due to the difference in definitions used to describe atypical features in the studies.Classical AVH begins with a prodrome that may include fever,nausea,vomiting,malaise followed by jaundice that resolves within the next 2-3 wk(Figure 3)[4].Any deviation from this classical presentation is included under the umbrella of atypical features.Prolonged cholestasis is the most common atypical feature that is seen in 11%,AVH with ascites in 7%,intravascular hemolysis in 3% and relapsing jaundice in 2%[3].
Prolonged cholestasis
Natural history:In the natural history of typical AVH,the cholestatic phase lasts for a brief period(3-4 wk)[5].Prolonged cholestasis is an atypical manifestation that is worrisome for the caregivers,a dilemma for physicians and troublesome for symptomatic patients.Deepening jaundice,pale stools,intractable pruritus,multivitamin deficiency features,fatigue,poor quality of life,school absenteeism and psychosocial difficulties predominate in this phase.In developing countries,the problem is compounded by self-imposed dietary restrictions that perpetuate malnutrition.In the initial description of cholestasis in patients with HAV infection,symptoms lasted for > 12 wk typically with serum bilirubin > 10 mg/dL and aminotransferases < 500 IU/L[6].Liver biopsy in patients with cholestasis showed portal inflammation rich in plasma cells,centrilobular cholestasis,ductular proliferation and cholestatic rosettes.Periportal necrosis may impede the normal bile flow contributing to cholestasis[7].In a pediatric study of prolonged cholestasis in HAV infection,the initial liver biopsies showed extensive periportal and centrilobular fibrosis similar to chronic hepatitis.A repeat biopsy 3 mo after resolution of cholestasis in the above cohort showed complete normalization and resolution of fibrosis[8].Children who develop this deviation from the classical presentation are older(> 10 years)and more commonly have HAV(15%)as compared to HBV(1%),HEV(9%)and coinfections(11%).Cholestasis can last up to 6 mo and 10% can also have relapsing hepatitis.Jaundice to pruritus interval is usually 20 (15-30)d[3].Figure 4 depicts the natural history of the prolonged cholestatic phase.It is postulated that HAV infection triggers polymorphisms in ATP binding cassette proteins that are involved in bile secretion[9].It has also been observed that in prolonged cholestasis,the time taken for HAV RNA to become undetectable in the serum is 46-105 d as compared to 11-20 d in classical hepatitis[10].These findings indirectly indicate that a complex interaction exists between genetic predisposition,immune response to HAV and prolongation of cholestasis.
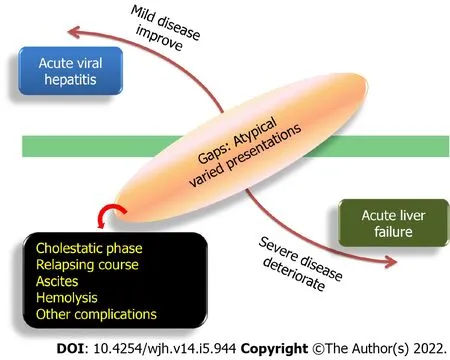
Figure 1 Spectrum of acute viral hepatitis.

Figure 2 Overlapping atypical manifestations of acute viral hepatitis.CLD: Chronic liver disease;DILI: Drug-induced liver injury.
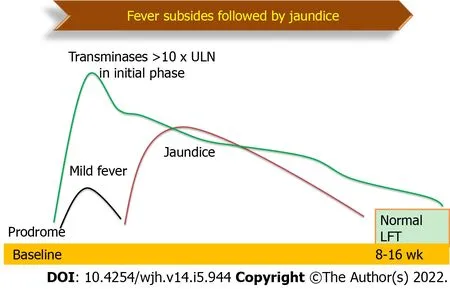
Figure 3 Natural history of classical acute viral hepatitis.LFT: Liver function tests.
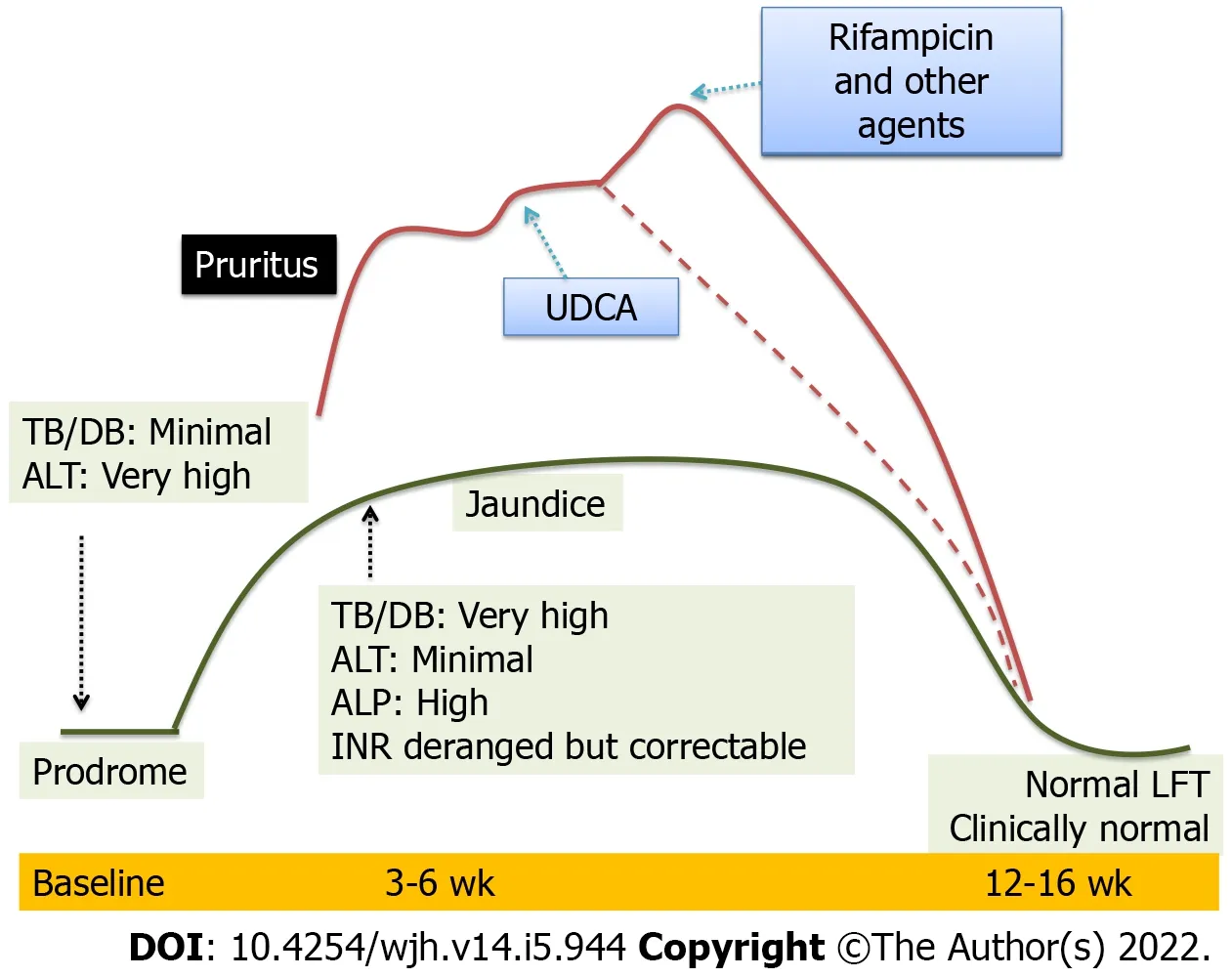
Figure 4 Natural history of prolonged cholestasis in acute viral hepatitis.LFT: Liver function tests;UDCA: Ursodeoxycholic acid;ALT: Alanine aminotransferase;TB/DB: Tuberculosis/Disulfide bond;ALP: Alkaline phosphatase;INR: International normalized ratio.
Differential diagnosis:When a child presents for the first time with a long history of icterus(progressive or relapsing),pruritus or pale stools,the tendency would be to look for cholestatic chronic liver diseases.Intrahepatic causes of cholestasis include progressive familial intrahepatic cholestasis,sclerosing cholangitis,Alagille syndrome,cystic fibrosis,alpha-1-antitrypsin deficiency and infiltrative disorders.Extrahepatic causes of cholestasis are choledochal cyst,Caroli’s disease,sclerosing cholangitis,biliary stricture,biliary compression by lymph nodes or masses and portal cavernoma cholangiopathy[11].Clues in the clinical presentation,extrahepatic manifestations,familial clustering,features of portal hypertension and radiology distinguish the above.
Mutations inATP8B1,ABCB11andTJP2present as cholestasis from early infancy with persistently low or normal -glutamyl transpeptidase(GGT)levels and liver dysfunction[12].Hence these disorders are rarely confused for AVH with prolonged cholestasis.Mutations inABCB4gene that result in a dysfunctional multidrug-resistant protein(MDR)3 may have milder phenotypes that appear for the first time as cholestasis during AVH.These disorders have a significant family history,maternal antenatal cholestasis,recurrent cholestatic pattern,biliary(intrahepatic and gall bladder)calculi and persistently high GGT levels[13].Liver biopsy would demonstrate periportal fibrosis and ductular proliferation with absence or faint staining of MDR3 protein in immunohistochemistry[14].Benign recurrent intrahepatic cholestasis is a milder variant in all the above disorders that have recurrent bouts of cholestasis in the second decade without progressive liver disease[15].These bouts may overlap with AVH.Genetic analysis should be sent if clinical suspicion is strong.
Alagille syndrome is an autosomal dominant disorder due to mutation in eitherJAGGED1orNOTCH2genes with incomplete penetrance and variable expressivity[16].Hepatic involvement may range from asymptomatic elevation of transaminases to decompensated CLD.The majority will have cholestasis in early infancy which can progress or resolve.A small proportion may present for the first time with late-onset cholestasis up to the age of 10 years who may mimic prolonged cholestasis of AVH[17].Characteristic facies along with positive family history,cardiac and skeletal abnormalities,will provide clues to the underlying disease.Liver biopsy showing paucity of interlobular bile ducts and/or positive mutation will confirm the diagnosis[18].
It is known that smooth muscle actin(SMA)and antinuclear(ANA)autoantibodies,may be incidentally detected during AVH due to autoimmune phenomena[19].Those presenting with cholestasis may be mistaken as having autoimmune sclerosing cholangitis or overlap syndrome.In those with extrahepatic autoimmunity,features of portal hypertension and biliary changes on sonography should be worked up with magnetic resonance cholangiography and/or liver biopsy[20].
Drug-induced liver injury(DILI)often complicates a regular course of AVH.A variety of complications such as a surge in transaminases,liver failure and cholestasis may occur during this period.It is important to carefully elicit a concomitant drug intake,especially herbal and alternative medicines.Liver tonics and immune boosters are marketed widely in Asia and obtained over the counter with a myth of hastening the recovery of icterus[21].Commonly used drugs that can cause cholestasis include antibiotics like amoxicillin-clavulanic acid,trimethoprim-sulfamethoxazole,erythromycin and tricyclic antidepressants[22].Temporal correlation with drug intake and improvement upon withdrawal supports the diagnosis of drug-induced cholestasis.However,in cases of chronic cholestasis,improvement may not be evident even after drug withdrawal and may progress to CLD[23].In Asian children with DILI,39% of cases were accounted for by herbal medications and the rest by antitubercular drugs,antibiotics and antiepileptics.Twenty-seven percent had a cholestatic pattern and 22% had a mixed pattern of injury based on the R factor.Fourteen percent went on to develop chronic DILI.Non-hepatocellular injury pattern was one of the factors predicting poor outcomes[24].
In a setting of prolonged cholestasis in AVH,an extensive workup including liver biopsy is generally not required.It is prudent to wait and watch for the natural resolution to take place if the liver functions are improving over 3-4 mo.Detailed workup for chronic diseases is indicated if there are significant clues in presentation,static or worsening liver dysfunction.
Treatment:Although spontaneous resolution can be seen,sequential pharmacotherapy may be required in those with significant and debilitating cholestasis-related pruritus.Ursodeoxycholic acid could aid in the resolution of cholestasis in 80% of cases.It increases the hydrophilicity of bile,induces the activity of carrier proteins for increasing biliary secretion and has antiapoptotic activity[25].Addition of rifampicin is required in 18% of cases.Rifampicin acts as an agonist of pregnane-X-receptor,which in turn mitigates bile acid toxicity.Cholestyramine,a bile acid sequestrant is required in 4% of cases[3].In refractory cases,opioid antagonist(naltrexone)or selective serotonin reuptake inhibitor(sertraline)is used for severe pruritus[26].The role of steroids in prolonged cholestasis of AVH is questionable.Few case series have shown a response of refractory pruritus to prednisolone(1 mg/kg/d)for 2 wk with tapering over 4-8 wk[27].Multivitamin supplementation is prudent.
Relapsing hepatitis
Natural history:Relapsing hepatitis is seen in 2-4% of children with AVH in whom it typically presents as a biphasic illness or rarely polyphasic relapses[6,8].In this entity,the second phase of hepatitis occurs after the resolution of the first phase and near-normalization of liver functions.The interval period between the two hepatitic phases may last for weeks to months[28].The second or subsequent phases may be icteric or anicteric.There may be a recurrence of prodrome.The peaks of liver enzymes may be variable or similar to the first episode.The subsequent phase may be similar,less severe or more severe than the first episode.Rarely,the relapsed phase may culminate in ALF or complicate into a cholestatic phase[29].Figure 5 depicts the natural history of relapsing hepatitis in AVH.Since liver function tests are measured at fixed time points of follow-up,anicteric relapses may often be missed.Hence the true prevalence of relapsing hepatitis remains undermined.During the relapses,HAV RNA can be detected in the serum and IgM HAV remains positive[11].Relapse is postulated to result from incomplete elimination of HAV in the initial phase.Recent mouse model studies have shown that anti-HAV IgA immune complexes reach the liver from the enterocytes by enterohepatic circulation.The process is continuous until IgG response is initiated.In those with inefficient IgG response,recurrent seeding of anti-HAV IgA immune complex to the liver can cause relapsing hepatitis[30].Treatment is supportive with close monitoring for progression to ALF.
Differential diagnosis:Relapsing jaundice is the presenting feature in 33% of children with autoimmune hepatitis that can last from 6 to 24 mo.Apart from icterus,presentations are fatigue,weight loss and anorexia.Absence of acute viral markers,presence of stigmata of CLD,decompensation or features of portal hypertension warrant workup for autoimmune hepatitis[31].
Sclerosing cholangitis can present with multiple relapses and spontaneous remissions.Magnetic resonance cholangiography and workup for underlying etiology confirm the diagnosis in the majority of cases.However,about 13% of cases of primary sclerosing cholangitis have exclusive small duct involvement that can be confirmed only by liver biopsy[32].Children with extrahepatic biliary obstruction have a prominent history of recurrent fever,cholangitis and absence of prodrome that differentiates from relapsing viral hepatitis.
Children with ATP8B1 and ABCB11 disease present with recurrent jaundice in 8% and recurrentpersistent jaundice in 23% of cases.Pruritus is a universal feature in them[33].Recurrent jaundice in ABCB4 disease is variable,dependent on the severity of mutation and often precipitated by drugs[34,35].Dubin-Johnson syndrome is a rare disorder of bilirubin excretion due to mutations inABCC2gene,with a characteristic black liver caused by lysosomal pigment accumulation.These patients can present with recurrent jaundice with elevated conjugated bilirubin from as early as 7 years of age[36].Hemolytic disorders and Gilbert syndrome clinically present with recurrent jaundice;however,they have unconjugated hyperbilirubinemia[37].These disorders have normal liver enzymes and synthetic functions.
AVH with ascites
Natural history:Ascites in children with AVH is present in 13% of cases;of which 38% are clinically detectable and have high serum ascites albumin gradient(> 1.1).Eleven percent have evidence of ascitic fluid infection(AFI).Associated pedal edema is present in 33% of cases and pleural effusion(hepatic hydrothorax)in 44%[38].Children who develop ascites in AVH are younger,with lower serum albumin,lower total protein and higher prothrombin time,signifying greater liver dysfunction than those with uncomplicated AVH.Features of portal hypertension(esophagogastric varices,portal hypertensive gastropathy,dilated portal vein,and abdominal portosystemic collaterals)or chronicity(shrunken liver,nodular surface,irregular margins or differential lobe hypertrophy)are not seen.Diuretics are required in 44% of cases.Unlike CLD,the ascites responds rapidly and completely by 8 wk without any recurrence.The resolution is paralleled with complete improvement in liver function and normalization of liver size(appropriate for age)[38].Figure 6 depicts the natural history of ascites in AVH.The mechanism of development of ascites was elegantly demonstrated in a study in which hepatic venous pressure gradient(HVPG)and liver histology were analyzed,which showed that liver cell dropout caused sinusoidal collapse that impeded intrahepatic blood flow resulting in portal hypertension.This finding was corroborated by the demonstration of higher HVPG(> 6mmHg)in those with ascites[39].
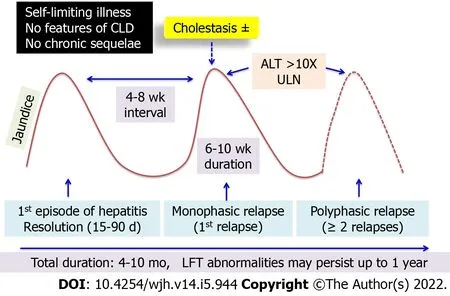
Figure 5 Natural history of relapsing hepatitis in acute viral hepatitis.CLD: Chronic liver disease;ALT: Alanine aminotransferase;LFT: Liver function tests.

Figure 6 Natural history of ascites in acute viral hepatitis.LVP: Levator veli palatine;SAAG: Serum-ascites albumin gradient;SBP: Systolic blood pressure;CLD: Chronic liver disease;LFT: Liver function tests.
Differential diagnosis:Ascites in the setting of liver dysfunction needs to be carefully analyzed.In a given case,there may be significant overlap between various entities of diagnostic and therapeutic dilemmas.Differential diagnoses of ascitic form of AVH are decompensated CLD,acute on chronic liver failure(ACLF)and LOHF as summarized in Table 1.Decompensation in those with CLD is defined as ascites,encephalopathy and/or gastrointestinal bleeding.Among all admitted children with CLD,40% have ascites[40,41].Twenty eight percent of children with decompensated CLD have evidence of AFI[42].According to the Asian Pacific Association for the Study of Liver Diseases(APASL),ACLF is defined as acute hepatic insult manifesting as jaundice(serum bilirubin > 5 mg/dL)and coagulopathy(INR > 1.5)complicated within 4 wk by clinically detectable ascites and/or hepatic encephalopathy in a patient with previously diagnosed or undiagnosed CLD[43].Among the children with ACLF,92% had ascites and 66% had hepatic encephalopathy.The majority of them presented for the first time with ACLF and CLD,and were silent until the precipitation of severe liver dysfunction.AVH was the precipitating factor in one-third of them,HAV and HEV in 35% each,HBV in 17% and Epstein-Barr virus in 11%[44].Thus,in a child presenting with jaundice and ascites,features of portal hypertension and chronic liver disease ought to be looked for actively.Esophagogastroduodenoscopy,portal vein diameter(age-appropriate cut-off values),HVPG and liver biopsy are required in selected cases.Other supportive elements such as presence of growth failure,stigmata of CLD and their etiologies,family history of CLD,or past history of similar symptoms reinforce the suspicion of an underlying CLD.

Table 1 Differentiation between Ascitic form of acute viral hepatitis,decompensated chronic liver disease,acute on chronic liver failure and late onset hepatic failure
Treatment:Restriction of dietary sodium to 2 mEq/kg/d,fluid restriction and diuretics form the mainstay of management of clinically significant ascites.Ascites in AVH rarely require aggressive management like large-volume paracentesis and albumin infusions.Diagnostic tapping of new-onset ascites in any liver disease is important for the presence of AFI.AFI needs to be treated as per the recommended antibiotic guidelines[45].
LOHF
Natural history:In the disease continuum of ALF,a subset of patients with liver dysfunction fall into the criteria where jaundice to encephalopathy interval ranges for a longer period commonly 5 to 12 wk.[46]This entity has been termed as sub-fulminant hepatic failure,sub-acute hepatic failure or LOHF.Other authors have re-defined the same entity differently in various studies with jaundice to encephalopathy interval ranging from 2 wk to 26 wk[47,48].Most experts agree that the cut-off between jaundice and encephalopathy should be > 4 wk interval to define LOHF.[49]The definition of pediatric ALF lacks a discrete timeline thus making it difficult to recognize LOHF in children[50].LOHF occupies a distinct position in the maze of severe liver dysfunction that is different from ALF and ACLF in terms of difference in its natural history and prognosis[51].
Of all the children with ALF,LOHF is seen in 8.3% with a median duration of illness of 53 d and an interval between jaundice and liver failure(ascites or encephalopathy)of 35 d.Ascites is the predominant sign of the failing liver seen in 94% and encephalopathy is present in 50%(advanced encephalopathy in 11%).Among all cases of LOHF,AVH is the most frequent etiology in nearly two-thirds with HAV being the most common which is also the predominant etiology of AVH(64%)as well as ALF(70%)in developing countries[6,52].In contrast HBV is the predominant etiology in adults with LOHF(32-46%)[53].Mortality is higher in children with indeterminate etiology,hepatic encephalopathy,renal failure,infection,coagulopathy and high pediatric end-stage liver disease(PELD)score.Thus,referral for liver transplantation should be considered earlier in this group of children with LOHF.PELD score > 32 can predict mortality or the need for liver transplantation with good sensitivity.Postmortem liver biopsy shows multiacinar massive or submassive necrosis without any features of chronicity.One-fourth of children recover spontaneously with native liver,as late as 24 mo after the onset of illness and without any evidence of CLD on follow-up[54].This is in contrast to adults who exhibit features of chronic hepatitis in LOHF at follow-up[51].It is imperative to recognize that LOHF is not merely old wine in a new bottle but is a distinctive entity whose natural history is yet to be
determined diligently in children.
Differential diagnosis:ACLF,decompensated CLD closely mimics LOHF.The absence of coagulopathy differentiates ascitic form of AVH from LOHF.
Treatment:Supportive treatment in lines of ALF management,maintenance of euglycemia,electrolytes,control of ascites and encephalopathy are required.Since they are at high risk of developing infections,a lower threshold has to be maintained for starting or changing antibiotics.Raised intracranial pressure is uncommon in LOHF thus,management of encephalopathy would be as per the guidelines for those with CLD[55].
Intravascular hemolysis in liver disease
Natural history:Intravascular hemolysis at presentation is encountered in 3% of children with AVH[3].They have severe anemia,deep icterus and dark-colored urine akin to cola.Since the jaundice is accentuated,there is often a dilemma in interpretation of liver function tests and chances of overdiagnosing the condition as terminal liver disease.Liver functions show a high fraction of unconjugated bilirubin.Aspartate aminotransferase is higher than alanine aminotransferase.Lactate dehydrogenase levels is markedly elevated.Reticulocyte count is high and peripheral smear shows schistocytes and hemolytic cells.Intravascular hemolysis is further evident by demonstrating plasma hemoglobin and urine hemosiderin elevation.Underlying glucose-6-phosphate dehydrogenase(G-6-PD)deficiency is often unveiled in an episode of AVH or by drugs like vitamin K.Among those with hemolysis,there is G-6-PD deficiency in 36%,direct Coomb’s test positive in 7%[3].It is crucial to identify hemolysis early to avoid drugs that precipitate red blood cell destruction especially vitamin K which is universally administered in AVH and also to start appropriate fluid management to prevent pigment nephropathy[56].G-6-PD levels may be falsely normal when measured during an acute episode of hemolysis due to relatively higher levels in new red blood cells.Hence,it has to be repeated after 3 mo to confirm G-6-PD deficiency,until then all contraindicated drugs have to be avoided.Figure 7 depicts the natural history of intravascular hemolysis associated with AVH.

Figure 7 Natural history of intravascular hemolysis(G6PD deficiency)in acute viral hepatitis.LDH: Lactate dehydrogenase;TB: Tuberculosis;IB: Illipe butter;DB: Disulfide bond;AST: Aspartate aminotransferase;ALT: Alanine aminotransferase;PRBC: Packed red blood cells;LFT: Liver function tests.
Differential diagnosis:Hemolysis is associated with CLD in Wilson disease and autoimmune hepatitis.Acute hemolysis as the presenting feature would be there in 6.7% of cases of Wilson disease[57].Acute release of copper from the necrosing liver cells is postulated to cause damage to the RBC membrane resulting in hemolysis,which is supported by finding very high serum and urine copper levels.Hemolysis is present in 22% of all children with Wilson disease and this proportion increases to 68% when considering those presenting as ALF[58].Leipzig score which is used to diagnose Wilson disease in children gives weightage to Coomb’s negative hemolytic anemia in addition to low ceruloplasmin,high urinary copper,high liver copper,positive copper staining on hepatocytes,Kayser-Fleisher ring,neuropsychiatric symptoms,positive family history and mutation in ATP7B gene[59].Acute hemolysis in Wilson disease is an emergency as it is an indication to start immediate plasmapheresis to remove excess serum copper and also list for liver transplantation[60].Autoimmune hemolytic anemia in children with autoimmune hepatitis is encountered in 35% of children.Coomb’s test will be positive and needs to be treated with steroids and/or immunoglobulins[61].
Treatment:In acute intravascular hemolysis in the background of AVH,with a presumed diagnosis of G-6-PD deficiency all contraindicated drugs should be stopped.Hyper-hydration and diuretics to maintain urine output would protect from pigment nephropathy[62].Simultaneous work-up for Wilson disease and autoimmune hepatitis ought to be done in select cases so that specific treatment is not delayed.In case of development of acute kidney injury,plasmapheresis and dialysis support may be required.
Autoimmune trigger
HAV infection itself can act as a trigger to uncover autoimmunity.In children with HAV,63% have positive autoantibodies(60% ASMA and 3% ANA).LKM antibodies are usually not found in concomitant AVH[63].The release of intracellular antigen during hepatocyte necrosis induces the antibody formation.The genes associated with the development of type 1 autoimmune hepatitis are located in HLA II DRB1 0301 Loci which are also reported to be present in those with persistent HAV infection.This may explain the precipitating role of HAV in autoimmune hepatitis[64].Though autoantibodies have been demonstrated in children with HAV,the exact role of HAV in triggering AIH is not well studied in children.
Systemic complications of AVH
Other than the above atypical features,systemic complications of AVH include acute kidney injury,thrombocytopenia,acute pancreatitis,hemophagocytic lymphohistiocytosis,aplastic anemia and transverse myelitis[65].
CONCLUSlON
Atypical presentations of AVH may have a myriad of presentations often mimicking an underlying CLD.Understanding the natural history of relapsing hepatitis,prolonged cholestasis,ascites and intravascular hemolysis in the setting of AVH aids in decision-making and avoiding unwarranted investigations that be may be unyielding.Invasive investigations and aggressive management must be carefully considered and indicated only when the suspicion is strong for an underlying CLD.LOHF is a distinct and aggressive variant of AVH that needs to be viewed in the lines of ALF.
FOOTNOTES
Author contributions:Sarma MS contributed the supervision and critical review of final draft of the manuscript;Ravindranath A contributed the literature search,data collection and writing first draft of manuscript;both authors wrote,read and approved the final manuscript.
Conflict-of-interest statement:Both authors have no conflicts of interests to disclose.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:India
ORClD number:Moinak Sen Sarma 0000-0003-2015-4069;Aathira Ravindranath 0000-0001-9323-7725.
S-Editor:Wang LL
L-Editor:Kerr C
P-Editor:Wang LL
杂志排行

World Journal of Hepatology的其它文章
- Role of hepatitis Β virus in development of hepatocellular carcinoma:Focus on covalently closed circular DNA
- Emerging curative-intent minimally-invasive therapies for hepatocellular carcinoma
- Saving time and effort: Βest practice for adapting existing patientreported outcome measures in hepatology
- Loco-regional treatment of hepatocellular carcinoma:Role of contrast-enhanced ultrasonography
- Βenign focal liver lesions:The role of magnetic resonance imaging
- Functions of three ubiquitin-conjugating enzyme 2 genes in hepatocellular carcinoma diagnosis and prognosis