氮掺杂锯齿型γ石墨二炔纳米带的构效关系研究
2022-07-02乔卫叶魏卫刚王莉莉张江玉赵新苗刘明娟
乔卫叶,李 敏,魏卫刚,王莉莉,张江玉,赵新苗,刘明娟
(邢台学院,河北邢台 054001)
一、前言
碳元素是构成人类生命的基础元素,碳也是地球上广泛存在的一种元素。碳有三种杂化形式:sp、sp2、sp3杂化[1],碳的杂化形式的多样性意味着碳可以形成不同的物质,这也就是我们所说的碳的同素异形体。近年来,国内外科学家们关于碳的同素异形体的研究一直方兴未艾,在其新的合成方法及其性能研究方面[2]收获颇丰:1985年,Rice大学和Sussex大学的Smally、Kroto和Curl等首次发现富勒烯;紧随其后的是1991年和2004年碳纳米管和石墨烯结构的问世;2010年在碳材料这一研究领域取得了突破性进展,中科院的化学家们制造出另一种新的碳材料——石墨炔[3],这成为了碳材料研究领域的热点话题[4]。
石墨炔是在石墨的基础上每两个碳之间连接两个炔键而形成的,具有良好的力学、催化和热稳定性等性能[5]。石墨炔的独特性能使其迅速发展成为一个新的领域,并且在燃料电池、金属空气电池等方面起着重要作用。陈彦焕等课题组在研究载流子运动过程时,运用氧化锌纳米粒子与石墨炔合成杂化纳米薄膜,提高了载流子的传输速率[6]。帅志刚等研究了石墨炔的电子结构,表示石墨炔的电子结构比较独特,而且该课题组通过计算形变势常数划分了石墨炔的适用范围,在此基础上指出了石墨炔的迁移率较高的特性[7]。神祥艳等对石墨炔的电化学性能作了详细的理论和实验研究,得出石墨炔基电池具有很好的储锂性能结论[8-9],并且在日常生活中石墨炔可以优异的容量存储性能广泛应用于超级电容器。尽管石墨烯与石墨炔都是2D离域π电子网络结构,但是石墨炔的异构体数目比石墨烯多得多,其结构具有更多未知性[10],因此也吸引了众多科学家参与到石墨炔这一新领域的研究中,从而在很大程度上实现了石墨炔的规模化、大范围的发展,为碳材料在日后的研究中带来了机遇。
值得一提的是,石墨炔具有高电荷传输能力,其常被应用在氧化还原反应中[11]。由于氮的尺寸与碳相差不大,但氮的电负性较大,吸引电子的能力也较强,故将氮原子引入到石墨炔中[12-14],可以增加邻位碳原子的正电性[15],增强其吸引带电体的能力,可以很好地提高反应活性[16]。石墨炔中掺入氮原子后,改变了石墨炔的一些物理化学性质,使其具有独特的性能,比如更强的活化性能和电学性能[17-20]。在酸性和碱性条件下,氮掺杂二维碳材料同样可以应用,石墨烯和石墨炔都表现出良好的催化稳定性[21-23],可以与常见的贵金属铂催化剂相媲美。将氮掺杂石墨炔运用到太阳能电池中,提高了能量转换效率和器件的短路电流值。
本论文针对近期合成的sp杂化氮掺杂石墨炔的新型材料,展开系统的理论研究。从原子水平上对新合成体系的结构参数,热力学稳定性,导电性能给予系统的探索。利用形变势理论,探索其载流子迁移率大小,分析伸缩模量,有效质量等对载流子速率的影响。
二、模型和计算方法
氮掺杂石墨二炔的实验合成路线如图1,可以得到α、β位置的氮取代结构。
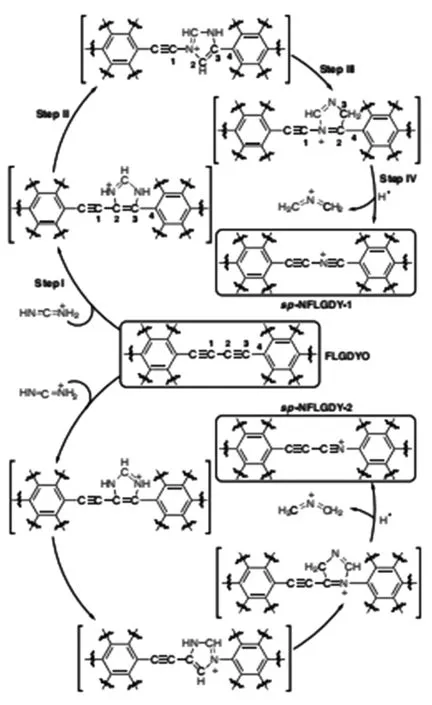
图1 氮掺杂石墨二炔的实验合成路线

图2 石墨二炔不同的氮掺杂方式
本模型选用的是二维的石墨二炔。图3为石墨二炔片层的结构,沿x轴方向将其剪切成一条一条的纳米带,由于剪切后的边缘为锯齿形,故称为锯齿形石墨二炔纳米带。

图3 石墨二炔结构

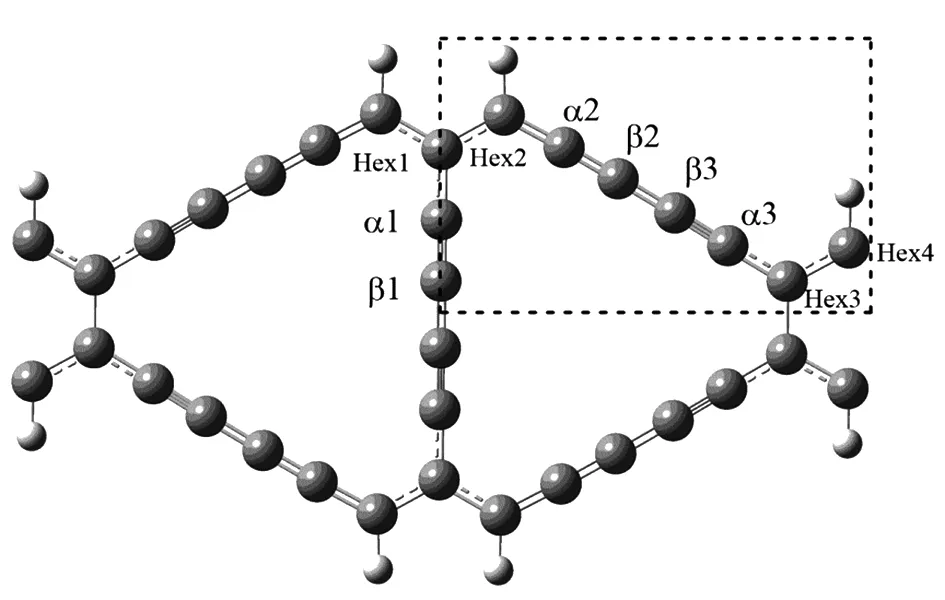
图4 锯齿型γ石墨二炔纳米带晶胞结构
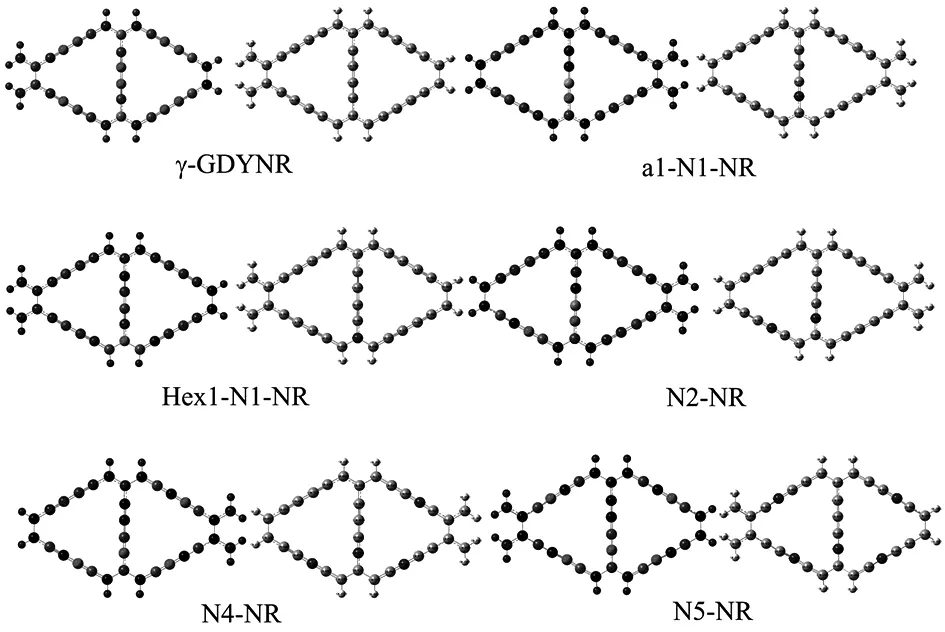
本文研究的是氮的掺杂位置、掺杂浓度对体系导电性质、力学性质、载流子迁移率、热力学稳定性等方面的影响。首先考虑在相同掺杂浓度下不同掺杂位置的影响:将单个氮原子掺杂到晶胞中,得出10个优化模型,图5为优化后平面图。其中六元环内的掺杂位置有4种,记为“Hex1-N1-NR、Hex2-N1-NR、Hex3-N1-NR、Hex4-N1-NR”;α碳原子上的掺杂位置有3种,记为“α1-N1-NR、α2-N1-NR、α3-N1-NR”;β碳原子上的掺杂位置有3种,记为“β1-N1-NR、β2-N1-NR、β3-N1-NR”,其中N1表示一个氮原子掺杂,Hex、α、β表示掺杂的位置。
接下来考虑的是不同浓度下的掺杂:两个氮原子的掺杂、三个氮原子的掺杂、四个氮原子的掺杂、五个氮原子的掺杂,并且得到了相关的优化模型,分别命名为:N2-NR、N3-NR、N4-NR、N5-NR。

图5 氮掺杂锯齿型γ石墨二炔纳米带平面图
图6为结构优化后单氮、二氮、三氮、四氮、五氮的侧面图,可以很直观的看出原子偏离平面的程度。
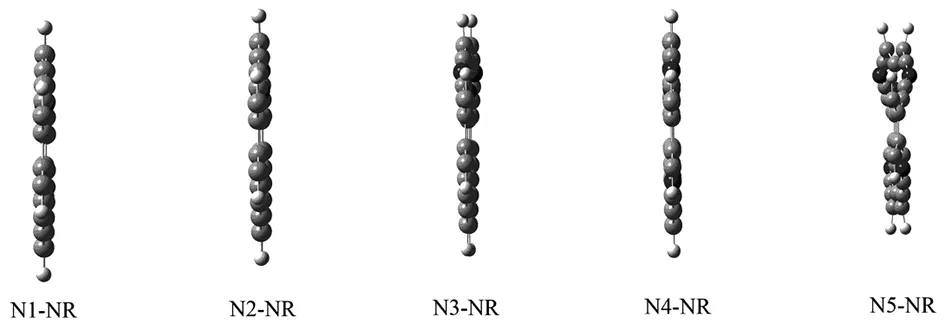
图6 氮掺杂锯齿型γ石墨二炔纳米带侧面图
本文计算所用软件均为专门用于固体计算的CRYSTAL 2014,采用密度泛函理论,相对于波函数理论,密度泛函理论可以在更高的精度条件下处理多原子的大体系,采用晶体轨道方法,交换相关泛函采用CRYSTAL 2014自带的专业分析固体的PBEsol 泛函,在半个布里渊区里,采用21个格矢点,收敛标准均采用程序的默认值。
三、结果与讨论
(一)结构参数和稳定性
由于本文的结构模型是在γ-石墨二炔的基础上进行局部的氮原子掺杂,当加入氮原子后,对原始结构的碳碳键长、结构的变形、偏离平面程度、结合能都会有影响。
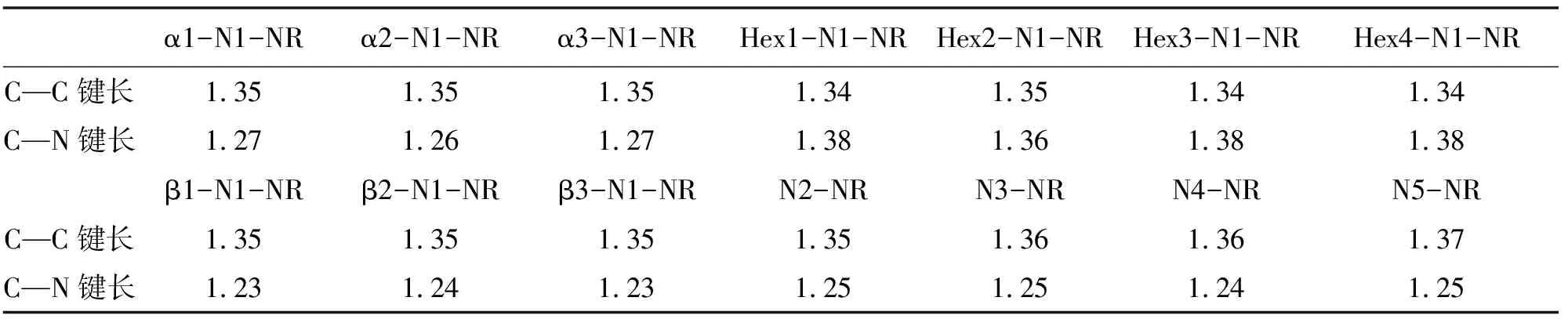
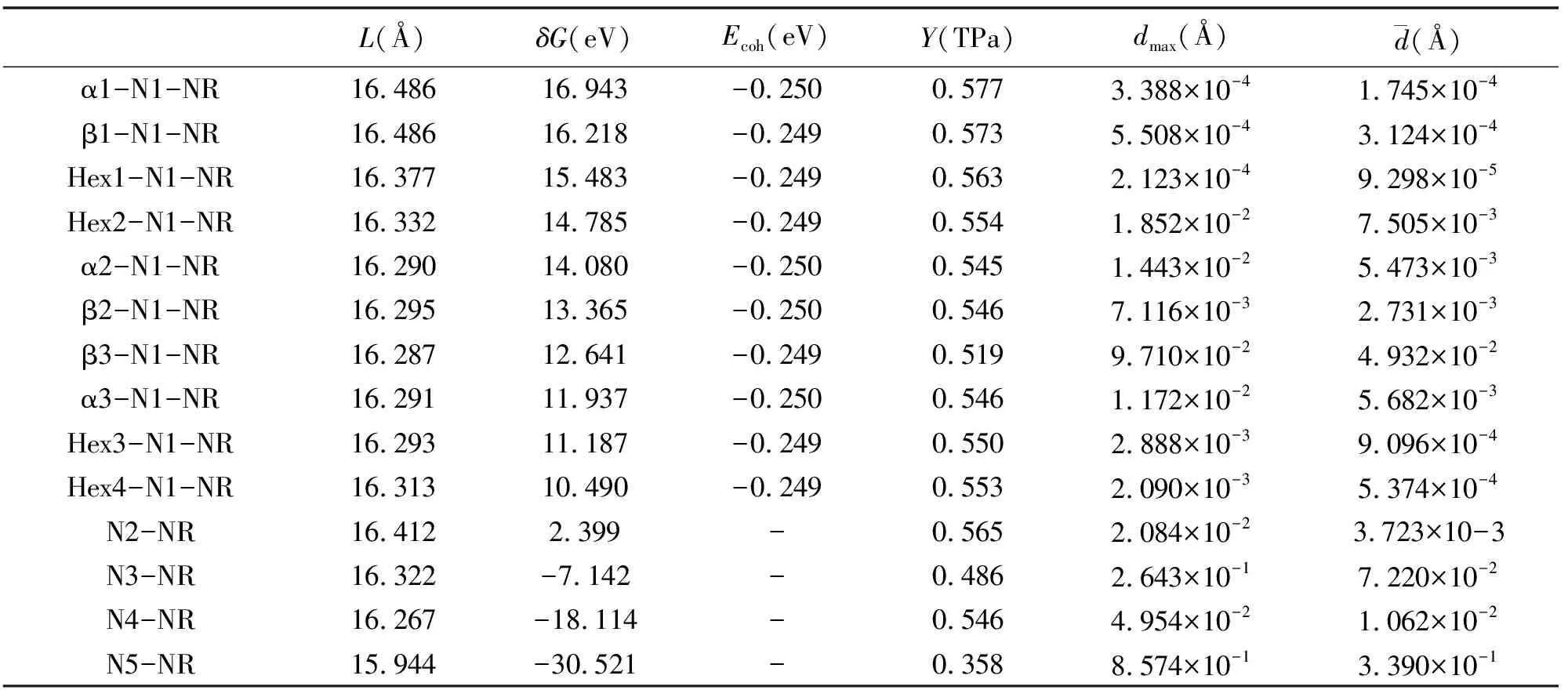
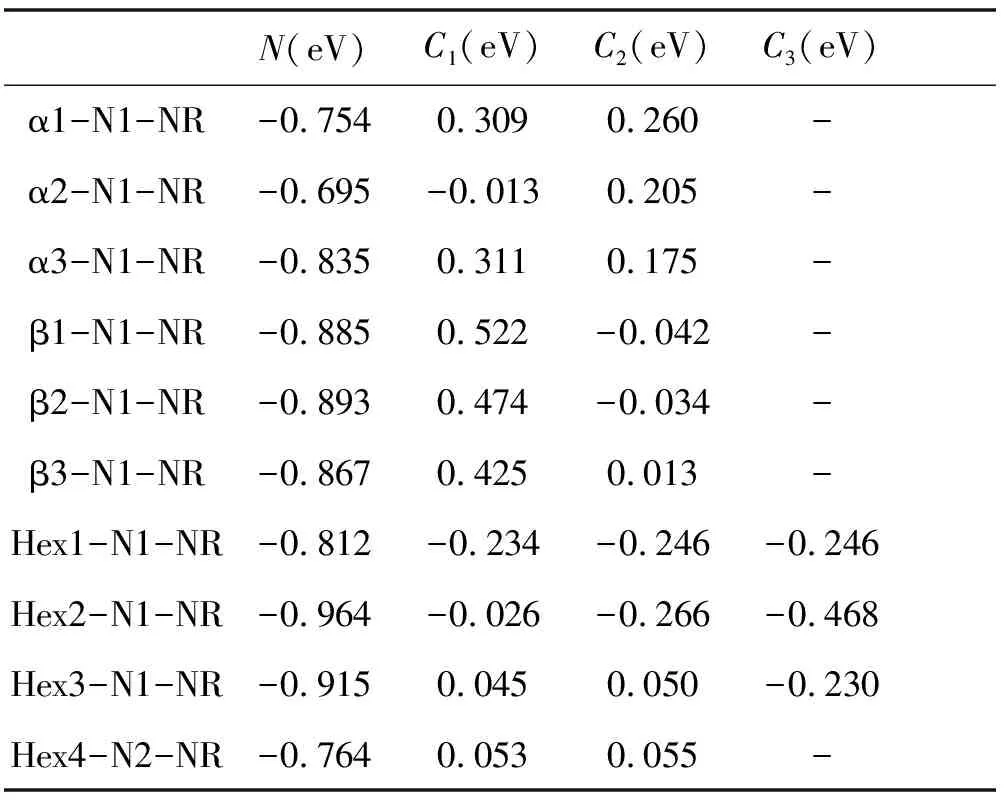
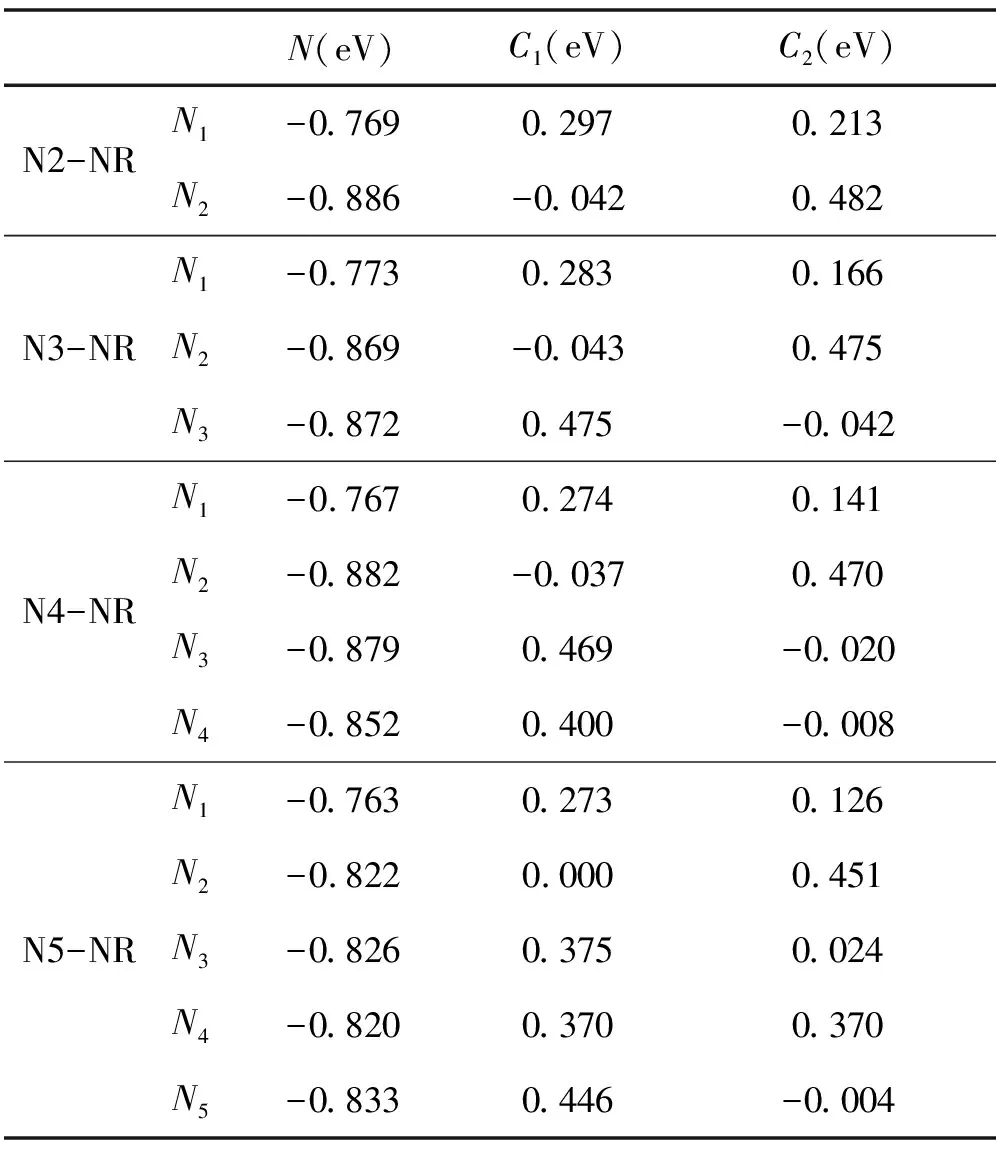
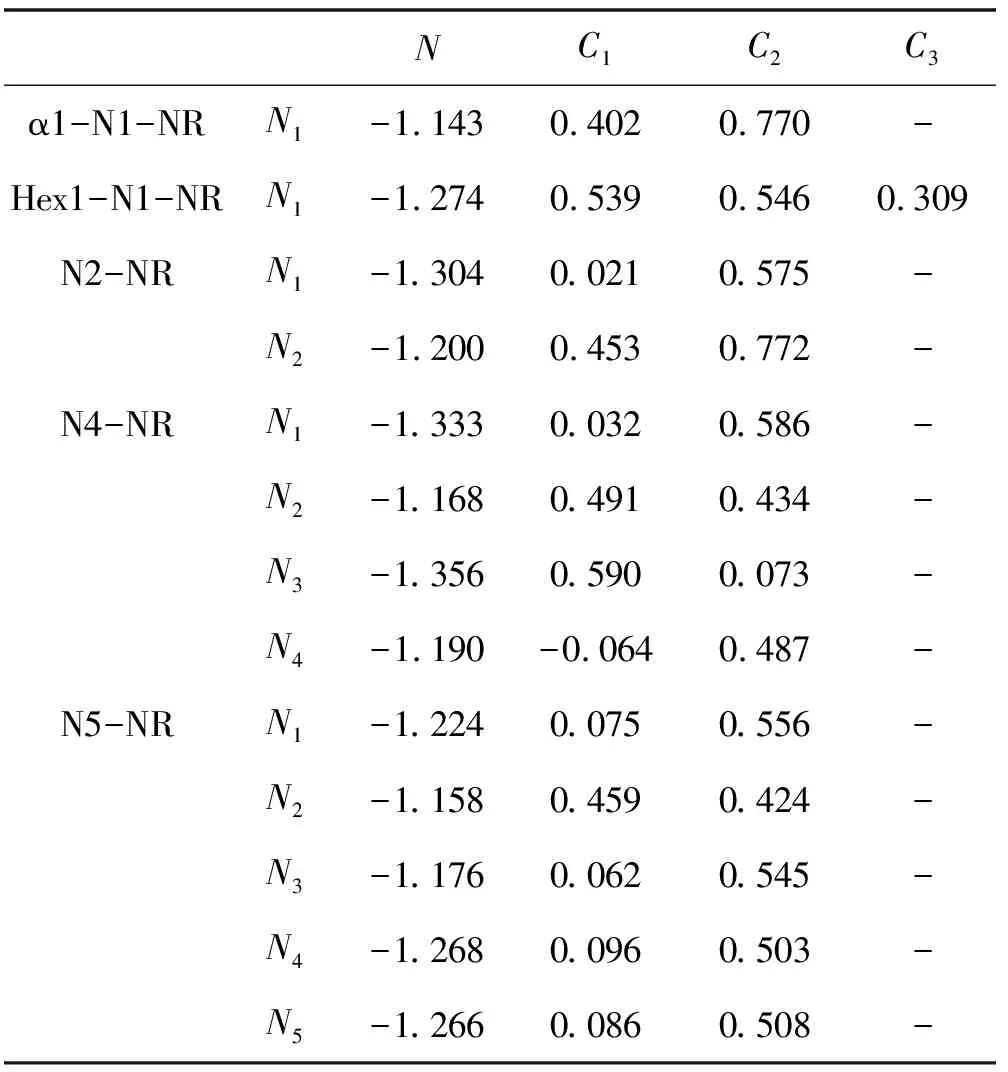
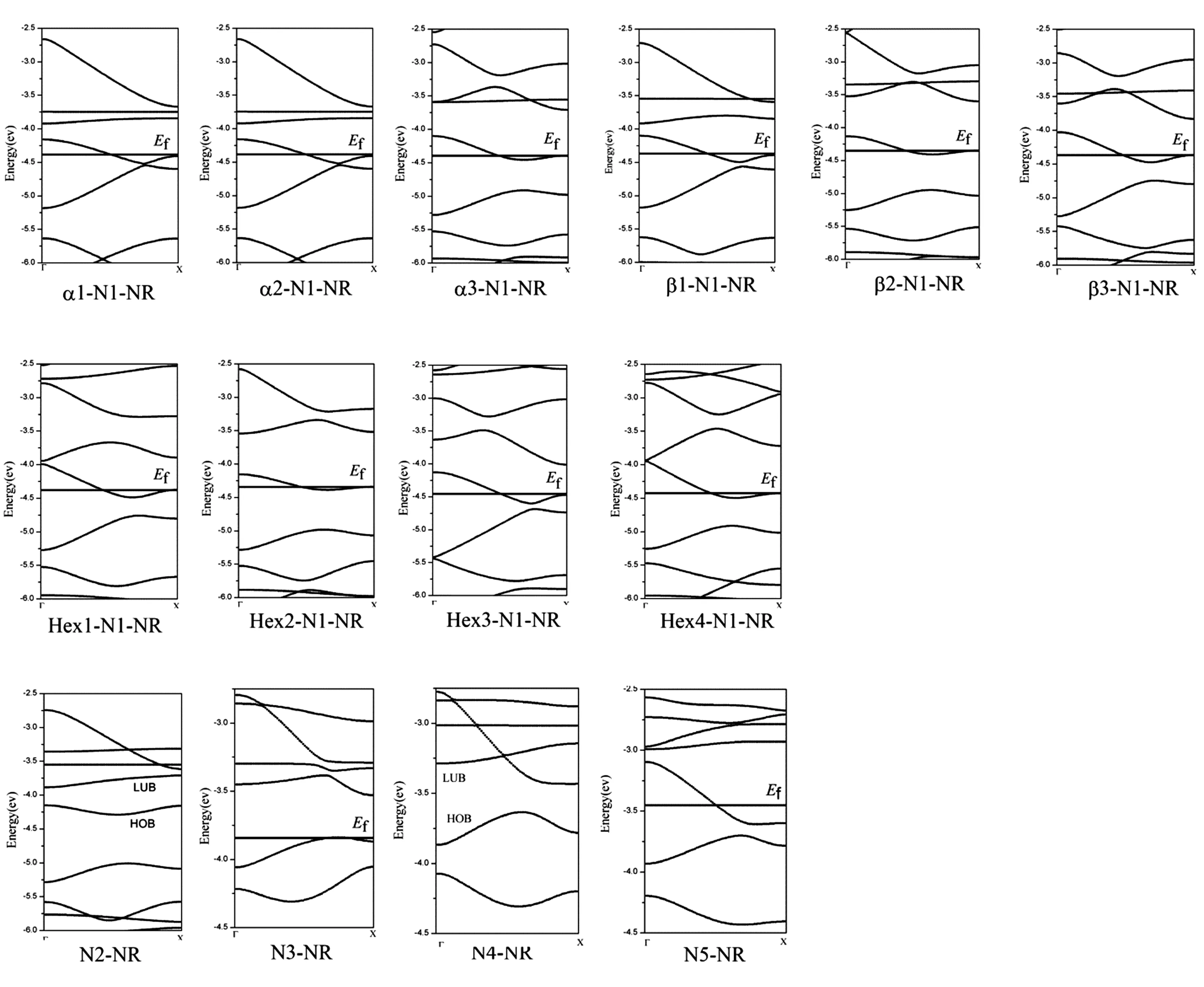
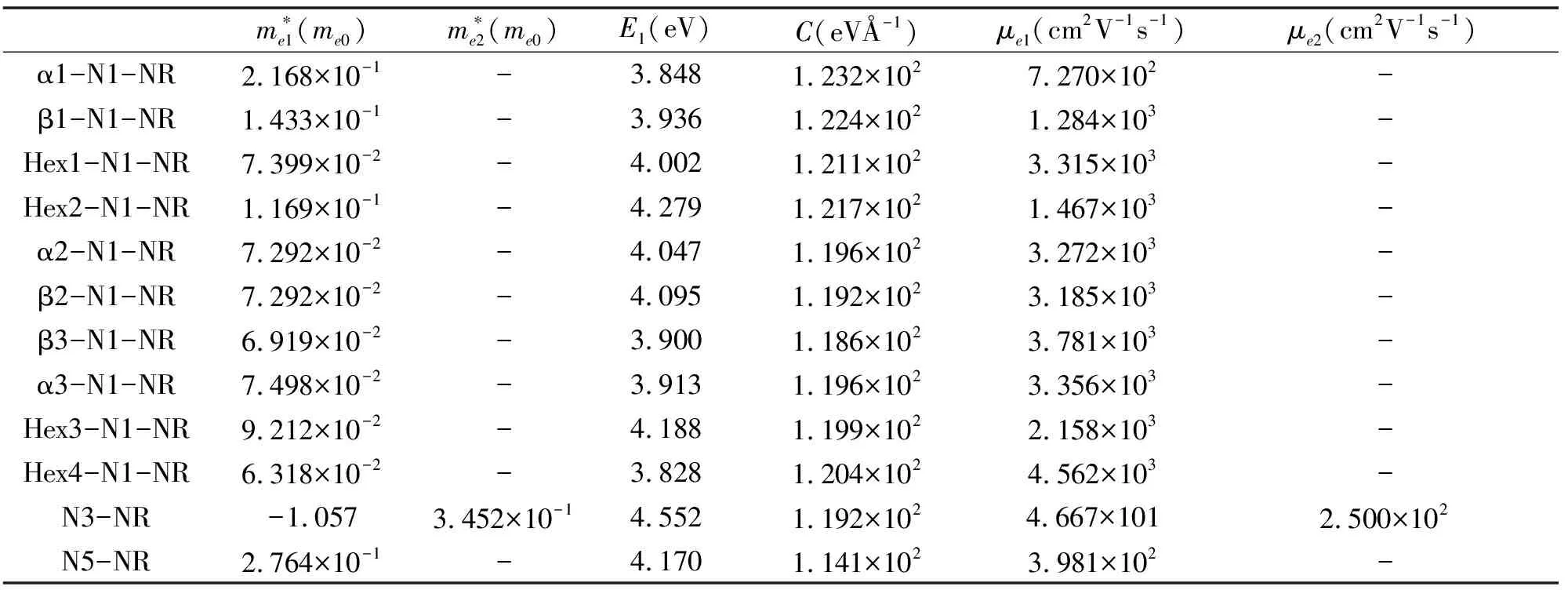
未掺杂之前石墨二炔纳米带中 碳碳键长范围为:1.234-1.464 Å之间,平均碳碳键长为1.349 Å,掺氮之后的碳碳键长,碳氮键长的范围在表1中给出,碳碳键长、碳氮键长的平均值在表2中给出,可以看出所有的碳碳键长的范围与原石墨二炔差别不大,所有单氮掺杂体系的碳碳平均值也变化不大,当氮的掺杂浓度大于等于3个氮原子每个晶胞后,由于局部张力造成的结构变形程度较大,碳碳键有略微增加的趋势。碳氮键的长度与氮原子掺杂位置之间有明显的关系,β位置掺杂碳氮键长为1.19-1.28 Å(平均值为1.23-1.24 Å),α位置掺杂碳氮键长为1.19-1.36 Å(平均值为1.26-1.27 Å),六元环内sp2杂化碳位置掺杂碳氮键长为1.32-1.43 Å(平均值为1.36-1.38 Å)。从平均键长和键长范围可以看出,碳氮键的变化规律为:β<α 表1 氮掺杂锯齿型γ石墨二炔纳米带碳碳键长、碳氮键长的范围(Å) 表2 氮掺杂锯齿型γ石墨二炔纳米带碳碳键长、碳氮键长的平均值(Å) 表3 氮掺杂锯齿型γ石墨二炔纳米带的晶胞参数、吉布斯自由能、内聚能、 杨氏模量、原子偏离平面的最大值和平均值 除了N5-NR 结构之外,其它结构的晶胞参数在16.2-16.5 Å之间,相差不大。对于N5-NR,由于结构偏离平面程度较大,造成晶胞长度要偏小。为了进一步衡量结构偏离平面的程度,给出了所有原子离开xy平面的最大距离dmax(Å)及平均距离,如表3所示。从总体趋势来看,对于掺杂浓度为 1(2)个氮原子每个晶胞的情况,偏离程度小于0.021 Å,而随着氮掺杂浓度增加,偏离程度越来越大,这可以从结构的侧面图直观的观察到。 为了衡量结构的热力学稳定性,可以用结合能Ecoh大小来表征,但结合能只可用来表征具有相同原子的数目和种类的不同体系的稳定性。然而,对于含不同原子个数或种类的纳米带结构,需要用每个原子的吉布斯自由能(δG)比较它们的相对热力学稳定性。δG越小,体系越稳定。 因为本模型里含有含奇数个电子的氮原子,所以按限制性空间和非限制性开壳层结构分别进行了计算,Crystal 2014 计算的结果显示,单个晶胞的能量差在0.01 eV以内,所以接下来的讨论都是基于限制性结构的结果。 本论文中有10个单氮掺杂体系,都含有38个原子,其中6个氢原子,1个氮原子,31个碳原子,这10个体系的稳定性比较可以用Ecoh,不同掺杂浓度体系稳定性比较需要用到δG。Ecoh和δG的定义式如下。 Ecoh=(E-nCEC-nNEN-nHEH)/n (1) δG=-Ecoh-χCμC-χHμH-χNμN (2) 其中μC,μN,μH分别表示碳原子、氮原子和氢原子的化学势,χC,χN,χH代表碳原子、氮原子和氢原子的摩尔分数。μC定义为石墨烯中碳的结合能,μN定义为氮分子中氮原子的结合能,而μH定义为氢分子中氢原子的结合能。 从表3中可以看出,单个氮原子掺杂体系的Ecoh差别不大,为-0.249到-0.250 eV之间,说明氮原子的掺杂位置对能量的影响较小。而对于不同掺杂浓度体系的稳定性比较,需要用到δG,计算的石墨二炔纳米带的δG为14.407 eV,通过比较,发现所有氮掺杂后的体系更稳定。从总体趋势来看,随着氮原子掺杂浓度的提高,δG越来越小,说明氮掺杂后,虽然有一定的变形程度,但是还是有利于体系的稳定的。 为了分析结构关键原子的电荷布局,用Crystal 2014计算的氮原子及其相邻碳原子的电荷数进行了统计,如表4所示,数值大小代表电子个数的多少,负值代表该原子得电子带负电荷,正值代表该原子失去电子带正电荷。 表4 单氮与相邻碳原子的电荷 表5 多氮与相邻碳原子的电荷 首先,对含有一个氮原子的10个体系进行分析。由于氮原子的电负性较大,所以计算结果表明,N原子均带负电,单氮取代体系氮原子得电子数值在 0.695-0.964 e 范围,而对于碳原子,在相邻氮的吸引下,大多数都是失电子而带有正电荷,范围集中在0.013-0.309 e内。但Crystal 2014 计算结果显示有部分氮邻碳原子依然带负电,这种情况存在于边缘六元环内碳原子被氮取代的情况(Hex1-N1-NR, Hex2-N1-NR),不符合常理,考虑是程序本身计算有误差,所以会和下边的Gaussian 09计算的结果进行对比。一维延伸方向晶胞连接处六元环内的氮取代碳原子电荷偏小。总体来看,单氮取代,3种β位置的取代造成的相邻碳原子的正电荷最大,其次是α位取代,而六元环位置的取代对增加相邻碳原子正电荷的效果最差。 图7是未掺杂氮原子的石墨二炔结构及部分掺杂结构的电荷分布图,分别给出每个原子上的电荷数以及正负电荷的分布情况,其中红色代表负电荷,红色越浅负电荷越大,绿色代表正电荷,同样绿色越浅正电荷越大,所以对于红色和绿色逐渐变深颜色的小球所带电荷都比较少。从图中可以看出,未掺杂石墨二炔结构中,所有β位置的sp杂化碳原子均带有负电荷,而α位置的sp杂化碳原子均带有明显的正电荷,与sp杂化碳原子相邻的六元环内的sp2杂化的碳原子带正电荷,而晶胞连接处六元环内不与sp杂化碳原子相邻的碳原子带负电荷,但从颜色上可以看出带有的负电荷量不大。 图7 石墨二炔掺氮前后电荷分布图 结合图7与表4、5可发现,当氮取代石墨二炔结构中原本带正电荷(Crystal 2014计算结果)的碳原子时,不会引起相邻碳原子带明显的正电荷,而当氮取代石墨二炔中原本带明显负电荷的β 碳原子时会引起相邻碳原子明显的正电性,有利于亲核反应的进行,而这对于催化活性的提高有明显的促进作用,对于晶胞连接带负电荷的碳被氮取代的情况,由于原本的碳原子所带负电荷并不多,所以相邻碳基本保持中性电荷状态(Hex4-N2-NR)。 N2-NR一个β位掺杂,一个ɑ位的掺杂,N3-NR位两个β位掺杂,一个ɑ位的掺杂,N4-NR 三个β位掺杂,一个ɑ位的掺杂,N5-NR四个β位掺杂,一个ɑ位的掺杂,由表6可看出,对于多氮掺杂系统,β位氮的临近碳均显示出明显的正电性,最少带0.273个正电荷(单位e),而所有ɑ位掺杂的相邻碳都接近中性,所带正电荷很少。 表6 Gaussian 09计算的代表结构的氮与 相邻碳原子的电荷(e) 上述表格(表6)是用Gaussian 09计算的电荷分布情况。选取了部分体系作为代表与Crystal 2014 计算结果进行对比。我们可以看出,Gaussian 09 计算结果显示氮原子带有更多的负电荷,而相邻的碳原子均带正电性,正电荷数值也比Crystal 2014结果要大。从电负性角度考虑,是更合理的。 从图7中可以看出,当引入氮原子后,原石墨二炔中β位碳原子带负电荷,ɑ位碳原子带正电性的分布会被打破,基本遵循着所有的两个sp2杂化碳原子间的电荷分布在没引入氮原子时基本保持原石墨二炔的分布,引入后其它三个碳原子都显示正电性或中性的分布规律。 计算结果显示锯齿形石墨二炔纳米带为导体,前线带为两条能带交叉。对非限制性体系的能带分别绘制ɑ自旋电子的能带和β自旋电子的能带图,经过对比后发现ɑ自旋和β自旋的电子能级完全一样,所以实际上体系是处于限制性状态的,所以图8给出的是限制性能带的体系,总电子对应的能带图。 在石墨二炔纳米带中对碳原子进行氮的取代属于n型掺杂,其中单氮取代的体系均为导体,图中用Ef标记费米能级的能级位置。每个晶胞2个氮,4个氮的掺杂浓度体系为半导体,在图中标出了最高占据晶体轨道(HOB)和最低未占据晶体轨道(LUB)。3个氮原子和5个氮原子体系为导体。通过对比氮掺杂体系的能带与石墨二炔相应的前线带对比,α1-N1-NR 与 α2-N1-NR 两个体系的原前线带还保持交叉,其它体系的这两条能带的形状均发生了很大的变化。总体上,奇数个氮原子掺杂体系均为导体,偶数氮原子掺杂的体系均为半导体,这点与总电子数为奇数的体系为导体,偶数为半导体相一致,也就是对导体或半导体进行n或者p型掺杂时是可以通过调整合适的掺杂浓度以期得到想要的导电性材料。 图8 单氮、多氮的限制性电子结构的能带图 图9是绘制的态密度图(DOS),虚线表示费米能级。由能带图可以看出费米能级(导体),价带,导带(半导体)的带宽都较大,所以费米能级处的态密度都较小,小于18 statee. eV-1. cell , 较小的态密度说明该材料不太适合作为超导材料的备选。 图9 氮掺杂石墨二炔纳米带态密度图 杨氏模量的计算基于公式(3),式中的V0代表体系一个晶胞的体积大小,二次偏微分的E指的是变形后对应晶胞的单点能,而ε对应体系的变形程度,分别选取伸缩的程度为ε=0%, ±0.5%, ±1.0% 和 ±1.5%几个值,以不同变形结构的能量对变形参数求二次导数,再除以体积大小即可求出杨氏模量的大小,其中曲线二次函数拟合要求线性关系在99%以上, 所得结果在表9中列出。 (3) 李林蔚[24]采用相同的计算软件,方法,基组,交换相关泛函和收敛限制计算的相同的石墨二炔体系的杨氏模量值大约为0.55Tp,引入单氮原子后的杨氏模量值并没有明显的减小,同样掺杂为2、4个氮原子的体系杨氏模量也没有明显减小,而掺杂为3个,尤其是5个氮原子的体系降低较多。归其原因,考虑是3/5个氮原子掺杂体系的结构变形比较厉害,偏离平面程度较强,在一定程度上降低了碳碳键的强度。 为了计算一维半导体和导体的载流子迁移率速率的大小,利用形变势理论推导出的公式作以下的计算: (4) 公式中的C指体系的伸缩模量的大小,衡量当拉长或缩短体系时,抗拉伸所需要的外力的大小,伸缩模量值越大,证明在拉伸方向上的力学性能越优良,计算公式为C=a0∂2E/∂a2|a=a0。me* 和mh*分别指体系电子和空穴的有效质量的大小,通常是相对于自由电子的数值,计算公式为m*=ħ2⎣∂2E/∂k2」-1。对于导体,需要计算伸长缩短对费米能级造成的影响,称之为形变势常数,用符号E1表示,而对于半导体需要分析价带顶和导带底随变形程度的变化率,需要计算E1c和E1v。默认的载流子迁移率的计算所处的温度为室温,也就是公式里的T取298.15K。本文对在一维方向上的拉伸变形程度选取:0.985a0, 0.990a0, 0.995a0, 1.005a0, 1.010a0和 1.015a0,加上原始长度共六个点,对所有的模型的能量和性质进行计算,绘制能带图,拟合相应的一次,二次曲线,保证线性系数在99%以上的精确性。最后把所有的量带入公式,得出电子和空穴的迁移率大小。 表7 导体的伸缩模量,形变势常数,有效质量,电子的迁移率 表7是本论文的所有导体的电子迁移率的计算结果,根据与费米能级相交的点个数,分别计算电子的迁移率大小。其中的有效质量是与自由电子的相对值。所有单氮掺杂体系的电子迁移率大小范围为:7.270×102-4.562×103cm2V-1s-1,相差不大。通过分析,这十个单氮原子掺杂体系的形变势,伸缩模量和有效质量差别都不是太大,其中α1-N1-NR的有效质量偏大点,造成了该体系的电子迁移率偏小些。 表8 半导体的伸缩模量,形变势常数,有效质量,电子和空穴的迁移率 表8是半导体结构的电子和空穴的迁移率相关数据。比较掺杂浓度的影响,发现多氮掺杂体系的迁移率相对单氮掺杂有所降低,这主要归结于局部的氮掺杂破坏了原碳原子体系的共平面共轭体系,包括氮原子与碳原子的体积差别,电荷排布差别,也包括多氮体系偏离平面的结构变形因素进一步的对共价p轨道的平行性造成影响。伸缩模量的变化趋势基本与杨氏模量值一样,单氮大,多氮降低,但N4-NR 变化处出现凸越,是因为相对于N3-NR和N5-NR,N4-NR原子的共平面效果要好一点。总体上,所有氮掺杂的石墨二炔体系依然能保持较为良好的载流子迁移率状态。 石墨炔作为一种新型的碳纳米材料,有着独特的性能,通过氮掺杂是改变石墨炔物理性质的重要方法之一。在结构和热力学稳定性上,随着氮掺杂浓度的提高,偏离平面的程度越来越大,δG越来越小,说明虽然高的掺杂浓度造成了一定的变形程度,但还是有利于体系稳定的。低浓度掺杂可以保持平面性质,对杨氏模量影响不大,然而多氮掺杂破坏了原有的共轭结构,力学性能有所下降。在电子能带研究中,证明奇数个氮原子掺杂体系均为导体,偶数个氮原子掺杂体系均为半导体。DOS图中费米能级处较小的态密度说明该材料不太适合作为超导材料的备选。在比较掺杂浓度的影响时,发现多氮掺杂体系的迁移率相对单氮掺杂有所降低,但是总体上,所有氮掺杂的石墨二炔体系依然能保持较为良好的载流子迁移率状态。如今,很多研究人员已经投入到石墨炔这一研究领域中,很大程度上改善了氮掺杂石墨炔的性能,然而,目前对氮掺杂石墨炔的研究尚不完善,希望未来可以探索出低成本高质量的制备方法,将石墨炔的优异性能充分发挥出来。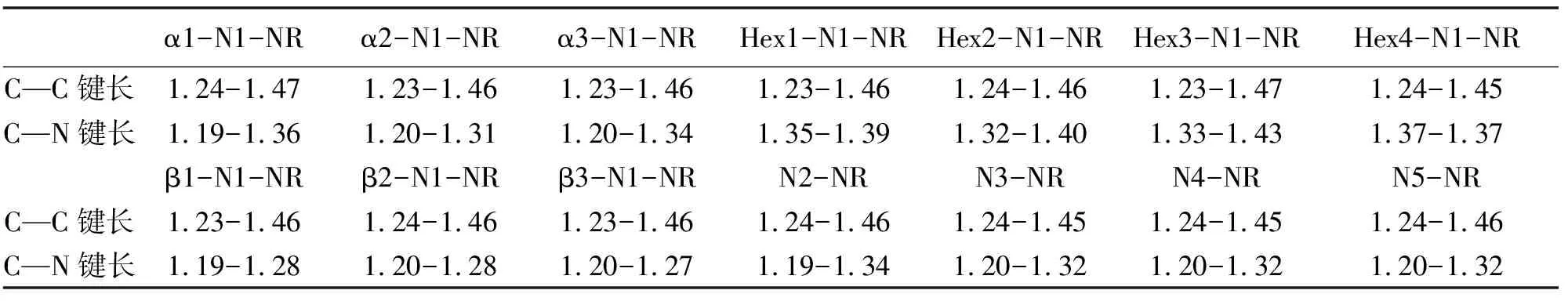
(二)电荷分布
(三)电子性质
(四)杨氏模量
(五)迁移率

四、结论
