食用油中多环芳烃检测的前处理方法研究进展
2022-07-02孟祥茹胡乐乾尹春玲
孟祥茹,胡乐乾*,琚 荧,张 艳,尹春玲*
(河南工业大学化学化工学院,河南 郑州 450001)
食用油是人们膳食结构中的重要组成部分,可为人体提供必需脂肪酸、VE、植物甾醇等营养物质,在人们生活中发挥着重要的作用。然而近年来,食用油中多环芳烃的检出多有报道,主要污染来源有种子的污染、油料加工的污染、油脂生产工艺上的污染以及食品外包装的迁移污染。且有研究显示随着油脂煎炸时间的延长,多环芳烃及其衍生物的含量也有所增加。食用油脂是人体摄入多环芳烃的主要来源之一,以苯并[]芘为例,GB/T 2762ü2017《食品安全国家标准 食品中污染物限量》规定油脂及其制品中苯并[]芘最大限量为10 μg/kg。
多环芳烃是两个或两个以上的芳香族环的复杂化合物的总称,主要是由石油、煤、天然气、烟草、木料、烟熏食品和一些有机化合物热解或不完全燃烧产生的,在食品、水、土壤、灰尘等中广泛存在。多数多环芳烃不溶于水,易溶于有机溶剂,具有高亲脂性、高熔点和高沸点,同时也具有较强的生物富集性。它们的化学性质稳定、不宜降解,是环境的主要污染源之一。多环芳烃根据苯环的数目可以分为两类:轻质多环芳烃(4个或少于4个苯环)和重质多环芳烃(5个或更多苯环),通常重质多环芳烃要比轻质多环芳烃更稳定,毒性更强。研究表明,大多数的多环芳烃具有致癌性、致畸性和致突变性,它们可以通过身体脂肪迅速分布到人体各种组织中。其中,一些多环芳烃的代谢物具有与细胞蛋白质和DNA结合的能力,具有毒性作用,能够对细胞造成损伤,从而导致突变和癌症。长期食用多环芳烃含量高的食用油会对人体造成一定的危害,因此对食用油中的多环芳烃进行检测十分重要,其检测方法对于鉴定食用油品质具有重要影响。
食用油中多环芳烃的检测主要包括样品的前处理和测定两个基本过程,其中常用的测定方法有气相色谱法、高效液相色谱法、超高效液相食用油中多环芳烃色谱法、表面增强拉曼光谱法、荧光分光光度法、色谱联用技术等。虽然检测方法众多,但检测仪器对样品要求较高,检测前大多都需要对样品进行前处理(除杂纯化富集多环芳烃),由于食用油中脂肪含量高,脂质基质复杂,且多环芳烃具有高亲脂性,前处理过程对于分析食用油中的多环芳烃是一个非常关键的过程,但目前仍然是一个很大的挑战,因此寻找合适的预处理方法,开发快速准确的方法来检测食用油中的多环芳烃具有重要意义。本文重点对近年来食用油中常用多环芳烃检测的样品前处理技术进行总结,并比较了不同前处理方法的优劣,以期为建立更高效的油脂中多环芳烃检测的前处理方法提供参考。
1 食用油中多环芳烃的前处理技术
由于多环芳烃的亲脂性高,在提取多环芳烃时,通常食用油中脂类成分会一起被提取出来,这会对食用油中痕量多环芳烃的检测造成较大的干扰,对测定结果产生不利影响。为了更有效地提取和富集多环芳烃,同时去除甘油三酯、脂肪酸等脂类杂质,需要选择合适的处理方法对油脂样品进行预处理,达到消除基体干扰,提高检测灵敏度的目的。目前,食用油中多环芳烃检测的前处理方法可以分为分离和纯化两个过程,分离方法主要有皂化法、索氏提取法、液液萃取法、超声波辅助萃取法、微波辅助萃取法;纯化方法主要有柱层析法、固相萃取、固相微萃取、分散固相萃取法、磁性固相萃取、分子印迹萃取等。
1.1 分离方法
1.1.1 皂化法
皂化法是利用碱性物质与食品中的脂肪等杂质发生皂化反应,进而除去脂肪酸盐的一种处理方法。由于多环芳烃是亲脂性物质,采用皂化法可以除去较多的脂质,达到分离、纯化多环芳烃的目的。常见的皂化法处理是采用氢氧化钾的乙醇或甲醇溶液在回流条件下分离油脂样品中的多环芳烃。
Zachara等采用皂化法,用1.5 mol/L的氢氧化钾-甲醇溶液回流90 min皂化植物油,并用环己烷进一步萃取,经浓缩、氧化铝柱净化,采用高效液相色谱-荧光检测器法(high performance liquid chromatography-fluorescence detection,HPLC-FLD)检测植物油中的4种多环芳烃,检出限为0.18 μg/kg,定量限为0.25 μg/kg,相关系数大于0.998,回收率为80%~110%。Akdoğan等采用皂化法,在60 ℃下用5 mol/L的氢氧化钾-乙醇溶液回流60 min皂化植物油,并用甲苯进一步萃取,经浓缩、硅胶固相萃取柱净化,采用HPLC-FLD检测植物油中的4种多环芳烃,在最优条件下,决定系数为0.998 0~0.999 9,检出限为0.06~0.12 μg/kg,定量限为0.13~0.24 μg/kg,回收率大于84.8%。Dost等采用皂化法,在60 ℃下用1 mol/L的氢氧化钠的甲醇-甲苯(体积比为1∶1)的混合溶液回流60 min皂化植物油,用甲苯进一步萃取,经浓缩、硅铝柱(质量比1∶1)净化,采用HPLC-紫外-可见检测器(ultraviolet-visible detector,UV-Vis)检测植物油中的9种多环芳烃,决定系数为0.995 1~0.999 6,检出限为0.26~1.15 μg/L,定量限为0.87~3.84 μg/L,回收率为80%~104%。此外,据报道一些学者认为苛刻的碱性处理可能会对样品中不稳定的多环芳烃产生影响。
在传统分离方法中一般会选择皂化法作为初步的前处理步骤,之后选择环己烷、二氯甲烷、,-二甲基甲酰胺(,-dimethylformamide,DMF)等溶剂进行进一步的液液萃取和纯化,以便分离油脂中多环芳烃。皂化法可以有效地去除食用油中大部分脂质,该方法简单廉价,适用于普通实验室。但是有机溶剂消耗大、耗时长,且仅用皂化法萃取会有杂质残留,必须进一步纯化,导致整个前处理过程繁琐耗时,不适合快速检测。
1.1.2 索氏提取法
索氏提取法也称作连续提取法,是一种传统、经典的前处理方法。王丽霞等采用自动索氏提取法提取,硅胶固相萃取柱净化,HPLC-UV-FLD法检测油炸面制品中的16种多环芳烃,该方法的相关系数大于0.999 0,回收率为92.5%~106.4%,回收率较高,但是耗费时间较长。Sun Ying等采用索氏提取法提取,并分别采用分子印迹固相萃取法和凝胶渗透色谱法两种方法净化,通过氧化铝固相萃取柱进一步纯化,并用气相色谱-质谱(gas chromatograph-mass spectrometer,GC-MS)法检测五花肉、黄油以及人脐带中的16种多环芳烃,结果表明分子印迹固相萃取法和凝胶渗透色谱法均能获得良好的回收率,除萘的回收率约为50%,其余多环芳烃的回收率均为75%~120%,结果较好。
索氏提取法经典方便,不需要复杂的仪器设备,回收率较好,但是有机溶剂需求大,操作繁琐、耗时,不适合快速检测,近几年使用较少。由此可见,开发快速、低成本、操作简便、绿色环保的前处理检测方法,并实现应用于油脂中多环芳烃的现场检测,是目前的研究热点。
1.1.3 液液萃取法
液液萃取是一种传统的样品前处理方法,利用目标分析物在两种互不相溶的有机溶剂中溶解度不同,从而使目标分析物转移到富集溶剂中,达到分离、富集目标分析物的目的。一般情况下,液液萃取需要进行多次萃取以提高回收率。食用油中多环芳烃传统的前处理过程是直接进行液液萃取或者先皂化处理后再进行液液萃取,萃取结束后采用柱层析法进行纯化,去除干扰物,富集多环芳烃。
Rascón等采用液液萃取法,将油样用正己烷溶解,然后用DMF-水(体积比为9∶1)的混合溶液萃取,之后运用以C为吸附剂的半自动固相萃取系统进行纯化,并利用GC-MS法测定橄榄油、芝麻油、大豆油等5种食用油中的16种多环芳烃,该方法的相关系数大于0.995,检出限为0.004~0.110 μg/kg,定量限为0.012~0.330 μg/kg,回收率为87%~104%,该方法灵敏度、选择性和精密度较高。Zhou Ruize等采用液液萃取法,将油样用己烷饱和的乙腈溶液萃取,并通过GC-MS法检测大豆油、花生油、橄榄油和玉米油中的多种环境污染物,其中23种多环芳烃的相关系数大于0.995,检出限为0.1~1.0 μg/kg,回收率为70.0%~110.8%,该方法避免了大量油脂和色素的干扰。Camargo等采用液液萃取法,将大豆油用己烷溶解,然后用DMF-水(体积比为9∶1)混合溶液萃取,C固相萃取柱进行纯化,并通过HPLC-FLD测定大豆油中的13种多环芳烃,该方法的决定系数大于0.999,检出限为0.02~0.76 μg/kg,定量限为0.03~0.96 μg/kg,回收率为71%~115%。Molle等通过同样的方法测定菜籽油、葵花籽油和玉米油中的13种多环芳烃,该方法的相关系数为0.993 3~0.998 9,检出限为0.07~0.30 μg/kg,定量限为0.3 μg/kg,回收率为71%~110%。然而该分析方法对苯并[]荧蒽(benzo[]fluoranthene,BjF)和茚并[1,2,3-,]芘(indeno[1,2,3-,]pyrene,IcdP)的灵敏度较低,这2种多环芳烃的检出限分别为1.95 μg/kg和1.32 μg/kg,定量限为3.0 μg/kg。Oh等采用液液萃取法,将油样用乙腈液液萃取,弗罗里硅土固相萃取柱净化,结合同位素稀释(isotope dilution,ID)-GC-MS测定食用油中的邻苯二甲酸盐、己二酸盐和多环芳烃,其中8种多环芳烃的决定系数为0.991 7~0.999 9,检出限为0.15~0.77 μg/kg,定量限为0.44~2.33 μg/kg,回收率为80.6%~96.9%。
作为一种传统的萃取方法,液液萃取的优点是操作简单、成本低廉、提取效率较好,但是易乳化,导致两相分离较慢,并且需要多次萃取分离来提高回收率,有机试剂消耗大,可能造成环境污染。综上所述,随着科学的发展以及人们对绿色化学的需求增加,选择体积小的萃取液进行提取,缩短相分离时间和前处理时间,提高萃取效率,实现微型化是当前液液萃取方法的一个研究趋势。
1.1.4 超声波辅助萃取法
超声波萃取法又称为超声辅助萃取法,其原理是通过超声波的空化作用和热作用,使多环芳烃快速有效地溶解到溶剂中。
Liu Yihong等以二甲基亚砜为溶剂进行超声辅助法萃取,通过同步荧光法检测植物油中的4种多环芳烃,该方法的检出限为0.16~13.00 μg/kg,回收率为69.3%~113.0%,该方法方便快捷、灵敏度高。Taghvaee等以乙腈和丙酮的混合溶液为溶剂,采用改良低温法和改良超声辅助液液萃取法,通过HPLC-FLD检测橄榄油和精炼果渣油中的15种多环芳烃。其中改良超声辅助液液萃取法分析时间更短,决定系数大于0.992 9,检出限为0.16~0.97 μg/kg,定量限为0.57~2.93 μg/kg,回收率为75%~111%。Wu Shimin等以正己烷为溶剂,利用DMF稀释,并进行超声辅助液液萃取,以弗罗里硅土固相萃取柱净化,通过GC-MS检测4种食用油中的16种多环芳烃,决定系数为0.998 0~0.999 9,回收率为70.11%~127.92%,该方法减少了样品和溶剂使用量。Zhao Weijun等以乙腈和丙酮的混合溶液为溶剂进行超声辅助液液萃取,以C固相萃取柱净化,通过HPLC-二极管阵列检测器(diode-array detector,DAD)-FLD检测大豆油、葵花籽油、玉米油等6种食用油中的16种多环芳烃,决定系数为0.998 8~0.999 9,检出限为0.01~2.35 μg/L,定量限为0.04~7.00 μg/L,回收率为57.3%~94.6%。
超声波萃取法简单、快速、便宜、溶剂用量少、萃取效率高,近几年在油脂多环芳烃的分离中得到了极大的应用,但在超声过程中易出现超声盲区,且超声时间过长,一些杂质会被共提出。因此,超声波萃取法一般需要多种前处理方法联用以去除共提杂质,同时应控制超声提取的能量和时间。减少超声盲区及避免共提取杂质的产生仍是该方法有待解决的问题。
1.1.5 微波辅助萃取法
微波辅助萃取法又称为微波辅助溶剂萃取,利用微波加热达到快速萃取样品中目标物的目的。微波加热的原理是基于偶极转动和微波对偶极子和带电分子或离子的离子传导效应。目前微波辅助萃取法在多环芳烃的萃取应用中比较多,但是在食用油中的应用较少。
Mohammadi等采用微波辅助分散液液微萃取法,结合GC-MS检测食用油中的14种多环芳烃,该方法具有快速、简便、溶剂消耗低、灵敏度高等优点,决定系数为0.934 6~0.997 8,检出限为0.2~2.7 μg/L,定量限为0.6~9.1 μg/L,回收率为84.4%~101.9%。Alarcón等采用微波辅助液液萃取,二氧化硅固相萃取柱净化,结合荧光分光光度法进行检测,采用偏最小二乘法(partial least squares regression,PLS)和展开偏最小二乘法(unfold partial least squares regression,U-PLS)算法预测初榨橄榄油和葵花籽油中7种重质多环芳烃的含量,该方法的相关系数大于0.994,检出限为0.8~7.0 μg/kg,定量限为2.4~22 μg/kg,且基于三维光谱数据的U-PLS算法,获得了最佳分析灵敏度、最低检出限和定量限,并通过HPLC-FLD对该方法进行评价,结果显示回收率为62%~84%;Alarcón等又采用相同的萃取方法,结合U-PLS/残差双线性(residual bilinearization,RBL)和平行因子分析(parallel factors analysis,PARAFAC)算法对三维荧光光谱进行分析,测定初榨橄榄油和葵花籽油中7种重质多环芳烃,其中U-PLS/RBL算法在此实验中更具优势,检出限为0.07~2.00 μg/kg,并采用HPLC-FLD法对该算法预测结果进行了评价,结果良好。
微波萃取法具有快速、环保、可批量处理样品、提取效率高等优点,但是不适用易挥发组分的提取,且能量过高可能会导致样品中其他成分也被提取出,因此需控制微波提取的能量和时间。此外微波萃取法需与其他样品前处理方法联用,以去除共提取物干扰。
1.2 纯化方法
1.2.1 柱层析法
柱层析法又称为柱色谱法,根据样品中各组分在固定相和流动相中的分配系数不同,经过多次分配,将不同组分分离。采用柱层析法分离纯化油脂中的多环芳烃时,固定相一般为中性氧化铝或者硅胶,流动相一般选择极性较弱的有机溶剂,如乙酸乙酯、石油醚、环己烷等。
Hossain等采用乙腈-丙酮(体积比为6∶4)混合溶液进行液液萃取,经过超声、离心、浓缩以及硅胶柱净化后,应用GC-MS检测大豆油、芥末油、椰子油中的8种多环芳烃,该方法的决定系数大于0.987,检出限为1.9~3.1 μg/kg,回收率为56%~84%。Zachara等采用皂化法进行分离,经液液萃取、浓缩后,采用氧化铝柱净化,结合HPLC-FLD检测植物油中的4种多环芳烃,相关系数大于0.998,检出限为0.18 μg/kg,定量限为0.25 μg/kg,回收率为80%~110%。Dost等采用皂化法进行分离,经液液萃取、浓缩后,采用硅铝层析柱(质量比为1∶1)净化,结合HPLC-UV-Vis检测植物油中的9种多环芳烃,决定系数为0.995 1~0.999 6,检出限为0.26~1.15 μg/L,定量限为0.87~3.84 μg/L,回收率为80%~104%。
柱层析法成本低廉、操作简单,但是耗时长、溶剂用量大、重复性较差,无法满足绿色环保的要求,近年来已经被固相萃取法取代。
1.2.2 固相萃取法
固相萃取是在柱层析基础上发展起来的一种样品前处理方法,原理是利用固体吸附剂吸附液体样品中的目标分析物,后续用洗脱液洗脱,达到分离、纯化和富集目标分析物的目的。固相萃取法目前已经被众多研究者用于食用油中的多环芳烃的分离纯化。固相萃取的大致操作流程如图1所示。

图1 固相萃取的操作示意图Fig.1 Schematic diagram of solid phase extraction
Shi Longkai等采用超声辅助液液萃取,硅胶固相萃取柱纯化,并通过GC-MS测定4种食用油脱臭馏出物中的16种多环芳烃,决定系数大于0.994,检出限为0.06~0.13 μg/kg,定量限为0.18~0.42 μg/kg,回收率为84.8%~115.5%。Lee等采用液液萃取,硅胶固相萃取柱纯化,并通过GC-MS测定芝麻油和红辣椒油中的4种多环芳烃,决定系数大于0.996,检出限为0.02~0.13 μg/kg,定量限为0.06~0.44 μg/kg,回收率为73.5%~112.6%,该方法时间较短,溶剂消耗少,背景基质干扰低,可以直接从油样中分离多环芳烃。Stenerson等采用含有C与ZrO-SiO混合物和活性弗罗里硅土的双层吸附固相萃取柱进行纯化,结合HPLC-FLD检测橄榄油中的16种多环芳烃,决定系数大于0.929,检出限为0.2~1.0 μg/kg,定量限为0.65~3.40 μg/kg,回收率为79%~127%。Ju等采用液液萃取,经超声、离心,运用EZ-POP NP双层固相萃取柱和氨基固相萃取柱纯化,充分去除了共萃取杂质,并结合ID-GC-高分辨率质谱(high resolution mass spectrometry,HRMS)检测橄榄油中的4种多环芳烃,检出限为0.08~0.10 μg/kg,定量限为0.10~0.28 μg/kg,回收率为97.5%~102%,结果可靠。
固相萃取法具有快速、准确、高效、有机溶剂用量少、选择性高、可自动化等优点,不足之处是不能重复使用,成本较高。目前,许多学者开发了很多新型的固相吸附剂材料,提高了固相萃取的萃取效率,使固相萃取的应用更加广泛,但更高效、更适用于油脂基质的高选择性固相吸附剂材料有待进一步研究。
1.2.3 固相微萃取
固相微萃取是在固相萃取基础上发展起来的一种新的前处理技术。固相微萃取是基于“相似相溶”的原理,利用待测组分在样品溶液或者顶空气体与萃取纤维涂层之间的分配平衡过程,达到萃取、富集的目的。根据测定的方式,固相微萃取技术可以分为3种:直接浸入式固相微萃取(direct extraction solid phase microextraction,DI-SPME)、顶空固相微萃取(headspace solid phase micro-extraction,HS-SPME)和中空纤维膜保护固相微萃取(hollow fiber membrane protected solid phase micro-extraction,HF-SPME)。以上3种固相微萃取方式的操作示意图如图2所示。

图2 3种固相微萃取方式操作示意图Fig.2 Schematic diagrams of three kinds of solid phase microextraction
Purcaro等采用DI-SPME技术,结合二维气相色谱-飞行时间质谱法,测定橄榄油、橄榄果渣油、葵花籽油以及植物油中的16种多环芳烃,决定系数大于0.957,检出限为0.2~1.4 μg/kg,定量限为0.4~4.6 μg/kg。此外,Purcaro等采用DI-SPME技术,结合GC-MS,测定橄榄油、橄榄果渣油、葡萄籽油以及4种谷物油中的10种多环芳烃,决定系数大于0.982,定量限为0.17~0.70 μg/kg,该方法减少了有机试剂消耗和甘油三酯的干扰,同时简化了实验操作。Chopra等通过HS-SPME技术结合GC-MS,测定鱼油中的12种多环芳烃,方法的决定系数大于0.990,检出限为1~7 μg/kg,定量限为3~21 μg/kg,回收率为80%~95%,该方法结果准确性较高,但在脂质复杂基质中平衡时间较慢。
固相微萃取灵敏度高,选择性好,与固相萃取相比,有机试剂的用量更少,操作更为简单,萃取富集的时间更短,回收率高,可实现固相萃取微型化,但是该方法的吸附容量有限,而且萃取头一般为石英纤维,易损坏,限制了其使用寿命,其较少应用于油脂中多环芳烃的前处理。因此,开发更高效、更适用于脂质基质的萃取涂层材料和研发新的固相微萃取装置解决萃取纤维头的使用寿命及成本问题是目前该方法的两个研究方向。
1.2.4 分散固相萃取
分散固相萃取法是在固相萃取基础上发展起来的一种新型前处理技术。分散固相萃取的吸附原理与固相萃取相似,区别是在分散固相萃取中,吸附剂不再以萃取柱形式存在,而是直接分散在样品萃取液中,通过振动、搅拌、超声等方式,使吸附剂与目标组分相互作用,在达到吸附平衡后,通过离心过滤使已吸附目标组分的吸附剂与样品基质分离,取上清液进行分析。固相萃取的操作如图3所示。
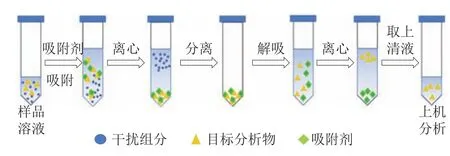
图3 分散固相萃取的操作示意图Fig.3 Schematic diagram of dispersive solid phase extraction
分散固相萃取最开始主要用于农药残留的提取,近些年在多环芳烃的检测中也有应用。Zacs等以多壁碳纳米管作为分散固相萃取吸附剂,对食用油样品进行预处理,并结合GC-MS,测定食用油样品中的4种多环芳烃,方法的决定系数大于0.983 3,检出限为0.06~0.21 μg/kg,定量限为0.19~0.71 μg/kg,回收率为96%~107%。Lv Zhiyang等以多孔金属有机骨架MIL-101(Cr)作为分散固相萃取吸附剂,对食用油样品进行处理,并结合HPLC-FLD,测定花生油、大豆油、菜籽油、辣椒油中苯并[]芘,该方法减少了有机溶剂的消耗,简化了操作,决定系数大于0.995 8,检出限为0.19 μg/L,定量限为0.56 μg/L,回收率为79.6%~117.1%。
分散固相萃取法简单快速、取样量少、溶剂用量小、成本较低、易于操作、选择性好,但是吸附容量有限,商业化和自动化程度较低。因此,需开发更高效、吸附容量更大、更适用于油脂基质的吸附剂材料,降低吸附剂材料的制备成本,研发分散固相萃取联用技术及装置,扩大其应用领域。
1.2.5 磁性固相萃取
磁性固相萃取是在分散固相萃取基础上,以磁性材料或可磁化材料为吸附剂的一种前处理技术。磁性固相萃取的原理是利用磁性或可磁性化吸附剂富集目标物,在外加磁场的作用下达到分离、纯化的目的。磁性固相萃取常用的材料有磁性碳基材料、离子液体、金属有机骨架、分子印迹聚合物等。磁性固相萃取的操作如图4所示。

图4 磁性固相萃取的操作示意图Fig.4 Schematic diagram of magnetic solid phase extraction
Wang Qing等以磁性多壁碳纳米管-十八烷基磷酸修饰氧化锆为磁性吸附剂,采用磁性固相萃取法结合HPLC-DAD,测定花生油、大豆油、葵花籽油中的6种多环芳烃,该方法高效、低成本、富集能力强、灵敏度高、可重复使用,决定系数为0.993 7~0.999 4,检出限为0.06~0.55 μg/kg,定量限为0.20~1.83 μg/kg,回收率为93.5%~113.2%。Zheng Haobo等以磁性氮化碳纳米薄片为磁性吸附剂,采用磁性固相萃取法结合GC-MS,测定大豆油、玉米油、茶油、葵花籽油中的8种多环芳烃,该方法的相关系数为0.996 7~0.999 8,检出限为0.1~0.3 μg/kg,定量限为0.4~0.9 μg/kg,其中在大豆油中回收率为91.0%~124.1%,其他3种食用油中回收率为79.1%~107.9%,该方法制备简单、重现性好、耗时短,只需要10 min。Ji Wenhua等以植酸稳定的氧化铁-石墨烯为磁性吸附剂,采用磁性固相萃取法,结合HPLC-DAD-UV-Vis,测定花生油、玉米油、大豆油和橄榄油中的8种多环芳烃,该方法的决定系数为0.998 9~0.999 9,检出限为0.06~0.15 μg/kg,定量限为0.2~0.5 μg/kg,回收率为85.6%~102.3%。Zhang Yun等以三维离子液体功能化磁性氧化石墨烯纳米复合材料为磁性吸附剂,采用磁性固相萃取法,结合GC-MS,测定花生油、大豆油、菜籽油、葵花籽油中的16种多环芳烃,该方法的决定系数大于0.999 2,检出限为0.05~0.30 μg/kg,定量限为0.17~1.00 μg/kg,该方法溶剂消耗少,可重复使用,易与样品溶液分离。
磁性固相萃取方法快速简单、绿色友好,极大地简化了前处理步骤,萃取速度快,便于回收,可重复使用,但尚未实现批量生产和自动化。今后的研究热点是开发稳定性好、选择性高、吸附容量大、富集能力强、萃取液使用量少、成本低的磁性吸附剂材料,推动其在食用油中多环芳烃检测方面的快速化、商业化和自动化发展。
1.2.6 分子印迹萃取
分子印迹萃取是一种高效的前处理方法,其原理是通过合成分子印迹聚合物,以其为吸附剂,利用分子印迹聚合物的构效预定性、特异识别性和广泛适用性,对目标组分进行吸附结合,达到分离纯化的目的。分子印迹固相萃取的操作示意图如图5所示。

图5 分子印迹固相萃取的操作示意图Fig.5 Schematic diagram of molecularly imprinted solid phase extraction
Zhou Hua等采用分子印迹固相萃取柱和多环芳烃专用固相萃取柱联用,分离纯化食用油中的24种多环芳烃,并通过GC-MS进行检测,该方法的相关系数大于0.999,检出限为0.1~1.0 μg/kg,回收率为86%~116%。Sun Ying等采用分子印迹多环芳烃专用固相萃取柱和凝胶渗透色谱法分离纯化脂质样品中的16种多环芳烃,并用氧化铝固相萃取柱进一步纯化,结合GC-MS检测,结果显示分子印迹萃取法与凝胶渗透色谱法测定的回收率一样高,且分子印迹萃取法的去除基质干扰效果更好,更有利于后续的GC-MS检测。Xu Ting等采用分子印迹多环芳烃专用固相萃取柱和活性碳固相萃取柱联用,分离纯化食用油中的24种多环芳烃,并进行GC-MS检测,该方法选择性、灵敏度高,决定系数大于0.998,检出限为0.03~0.60 μg/kg,定量限为0.13~2.00 μg/kg,回收率为56.8%~117.7%。Drabova等比较了凝胶渗透色谱、凝胶渗透色谱与硅胶固相萃取柱联用、多环芳烃分子印迹固相萃取专用柱3种方法纯化7种植物油中的16种多环芳烃的效果,其中多环芳烃分子印迹固相萃取专用柱的效果最好,定量限为0.1~0.3 μg/kg,回收率为70%~99%。
分子印迹技术能够对目标组分进行特异性识别,制备简单、溶剂消耗少、稳定性好、回收率高、检出限低,但在实际应用中仍存在几个问题:1)目前分子印迹技术多用于刚性小分子,且用于制备分子聚合物的一些模板分子和商品化的分子印迹固相萃取柱较贵,导致成本较高,未来可利用计算机模拟分子印迹聚合物的合成路线,节约制备成本,缩短制备时间,促进其商业化;2)分子印迹技术目前自动化程度较低,需加强分子印迹技术与检测技术的联用以促进其自动化。
1.3 分离、纯化相结合方法
1.3.1 凝胶渗透色谱
凝胶渗透色谱又称体积排阻色谱,是一种按分子尺寸大小的顺序进行分离的一种色谱分析方法,可根据样品中各组分分子质量的不同将其分离。由于油脂中主要成分甘油三酯的分子质量大于多环芳烃的分子质量,因此可用凝胶渗透色谱方法将多环芳烃从甘油三酯等基质中分离出来。
Wang等采用凝胶渗透色谱净化,结合UHPLCDAD-FLD测定食用油中14种多环芳烃,该方法的决定系数大于0.999,检出限为2.5~10.0 μg/kg,定量限为5~150 μg/kg,回收率为73%~110%。Zhu Huaming等通过环己烷-乙酸乙酯(体积比1∶1)混合溶液萃取,凝胶渗透色谱净化,结合HPLC-FLD测定茶油中15种多环芳烃,该方法的相关系数大于0.991,检出限为0.10~0.20 μg/kg,定量限为0.33~0.67 μg/kg,回收率为79.3%~97.3%。Wang Jianhua等以乙腈为溶剂,经过超声提取、自制凝胶渗透色谱柱净化,结合ID-GC-MS测定植物油中的16种多环芳烃,该方法的定量限为0.3~0.6 μg/kg,回收率为81%~96%,其自制的短窄凝胶渗透色谱柱可以显著减少溶剂和凝胶渗透色谱树脂的用量。Fromberg等采用凝胶渗透色谱和固相萃取技术相结合,并通过GC-MS测定食用油中24种多环芳烃,检出限为0.2~1.5 μg/kg,定量限为0.3~3.0 μg/kg,回收率为59%~120%。Ballesteros等采用液液萃取和凝胶渗透色谱净化相结合,结合GC-MS,同时测定初榨橄榄油、精制橄榄油和橄榄果榨油中的农药残留和4种多环芳烃,该方法净化效果好,其中多环芳烃测定的决定系数大于0.992,检出限为0.05~0.07 μg/kg,定量限为0.10~0.20 μg/kg,回收率为84%~110%。
凝胶渗透色谱将分离、纯化相结合,方法简单快速、易于操作、可自动化、回收率高、重复性好、稳定性好,被广泛应用于油脂中多环芳烃的分离纯化,但设备成本高、有机溶剂消耗大、耗时长。如何减少溶剂消耗、加快分析速率是目前凝胶渗透色谱亟待解决的问题。
1.3.2 QuEChERS前处理技术
Anastassiades等首次提出了QuEChERS方法,QuEChERS基于液液萃取和分散固相萃取的原理,利用吸附剂来富集目标物,提取目标组分,达到除杂净化的目的,是一种快速、简单、廉价、高效、安全的样品前处理技术。QuEChERS前处理技术的操作如图6所示。

图6 QuEChERS的操作示意图Fig.6 Schematic diagram of QuEChERS
QuEChERS技术最开始用于农药残留的检测,只能处理果蔬等基质较为简单的样品,而对于脂质含量较高的样品净化效果不太理想。处理油脂样品的关键是选择合适的溶剂和吸附剂,减少油中共提物的干扰,在QuEChERS方法中,常用的吸附剂有C、-丙基乙二胺(primary secondary amine,PSA)、石墨化炭黑(graphitized carbon black,GCB)、增强型基质去除(enhanced matrix removal,EMR)。
Sun Yaqing等采用EMR为吸附剂填料,利用乙腈-丙酮(体积比为3∶2)的混合溶液萃取,结合GC-三重四极杆(triple quadrupole,QqQ)-MS同时测定废弃煎炸油中的16种多环芳烃,该方法的决定系数大于0.995,检出限为0.06~0.13 μg/L,定量限为0.20~0.43 μg/L,回收率为66.72%~112.87%,提高了重质多环芳烃的回收率,减少了溶剂使用量,并实现了快速检测。朱捷等采用以PSA和C为吸附剂填料的QuEChERS方法,选择乙腈作为萃取试剂,结合GC-QqQ-MS同时检测枸杞籽油中16种多环芳烃,结果表明决定系数大于0.998,检出限为0.2~3.5 μg/kg,定量限为0.7~11.5 μg/kg,回收率为60.04%~119.00%,灵敏度高、结果较好。胡国绅等比较了以EMR吸附剂作为填料的QuEChERS法和凝胶渗透色谱法的分离效果,结合GC-MS测定植物油中的16种多环芳烃,与凝胶渗透色谱的净化效果相比较,QuEChERS测定结果的准确性和精密度较高,溶剂使用量较少,且决定系数大于0.995,定量限为0.3~0.8 μg/kg,回收率为88.9%~112.3%。
与其他方法相比,QuEChERS方法快速、简便、可靠、溶剂消耗少、样品制备相对便宜、回收率较为理想,是一种高通量的绿色前处理方法,但吸附剂的吸附容量有限。因此开发更高效的吸附剂材料、提高其吸附容量和重复使用次数仍是目前有待解决的问题,对进一步提高QuEChERS方法在快速分离富集食用油中多环芳烃方面的应用有重要意义。
除此之外,其他一些前处理方法,如加速溶剂萃取法、薄层色谱法、供体受体复合色谱、泡腾辅助微萃取、分散液液微萃取等也开始应用于食用油多环芳烃的分析中。
上述食用油中多环芳烃检测的样品前处理方法都具有较好的萃取效率和回收率,但是各前处理方法的萃取效果还是存在差异。表1总结了不同分离、纯化食用油中多环芳烃方法的优缺点。

表1 不同前处理方法提取多环芳烃的优缺点对比Table 1 Comparison of advantages and disadvantages of different pretreatment methods for PAHs extraction
2 结 语
本文重点综述了近年来食用油中多环芳烃检测过程中的样品前处理技术。传统的前处理方法如皂化法、索氏提取法、液液萃取法、柱层析法成本低廉,不需要复杂的设备,但是处理时间长、有机溶剂使用量大、回收率低、稳定性差,越来越难满足目前检测的需求。新型的前处理技术如固相微萃取技术、分散固相萃取技术、分子印迹萃取技术、磁性固相萃取技术快速简单、灵敏度高、溶剂使用量少、回收率高,但是吸附容量有限,且自动化和微型化程度低。虽然新型的萃取技术已被应用于从食用油中分离纯化多环芳烃,但更高效、快速、绿色、高通量、能够实现现场便携式检测的前处理技术仍有待开发。
建立方便、可靠、灵敏、快速、绿色无污染的前处理方法,实现设备自动化、小型化和便携化,始终是食用油中多环芳烃的检测前处理方法的研究趋势。未来食用油中多环芳烃检测前处理方法的研究可在以下几个方面实现突破:采用多种前处理方法联用,提高分离纯化的效率;开发新型的、吸附量高、更适用于脂质基质的吸附剂材料,提高灵敏度,促进商业化;开发新的前处理方法,简化前处理过程,实现自动化、小型化、便携化,促进其在现场快速检测中的应用。
