高效液相色谱-串联质谱法快速测定麻杏石甘口服液中喹诺酮类、磺胺类等17种药物
2022-07-01张培训张秋娜杨彦超
张 娜,陈 静, 张培训,张秋娜,杨彦超, 辛 雪
(1.保定冀中药业有限公司,河北保定 071000; 2.河北省中兽药技术创新中心,河北保定 071500;3.华谱科仪(大连)科技有限公司,辽宁大连 116000)
随着农业农村部减抗政策的实施,中兽药制剂因其低毒副作用、不产生耐药性更为广大养殖户所青睐,但同时伴随着国家监管力度的增加[1-5],在市场抽检过程中检出多种非法添加、残留药物,涉及磺胺类、喹诺酮类、四环素类、酰胺醇类、呋喃类等,使得养殖集团不得不从源头加强食品动物的用药安全把控,要求兽药生产企业按照动物源性产品中禁限用兽药的检测限来提供产品的检测结果,这对兽药生产企业来说是严峻的挑战。
但是从目前已颁布的多项中华人民共和国农业部公告及国家标准来看,对中兽药制剂中禁限用兽药的检测方法多为HPLC-PDA法,检测限难以满足养殖集团的用药需求,同时我们也发现公告及文献报道[6-10]中涉及的兽药制剂多集中于中兽药注射液、固体散剂、口服液剂型,前处理方法多为提取后直接稀释进样,这直接导致了检测的灵敏度降低,同时易污染设备。为降低中兽药中复杂的基质干扰,提高检测限,宋军、马东杰等[11-12]采用PRIME HLB对基质进行净化,检测中兽药中硝基咪唑类和硝基呋喃类药物,使检出限和定量限低至10~25 μg/kg,但对于养殖集团关切的多种禁限用兽药的检测未有提及。
因此,建立一种快速、简便、灵敏、可靠、可同时检测中兽药中多类非法添加或药物残留的禁限用兽药的检测方法十分必要。本实验室首次将亲水亲油平衡材料(HLB SPE)固相萃取柱用于麻杏石甘口服液的前处理,经考察17种禁限用兽药的回收率均符合要求。其中喹乙醇的检出限为10 ng/mL、定量限为25 μg/mL,剩余16种药物的检出限均低至5 ng/mL、定量限为10 ng/mL,极大保障了中兽药产品用药安全,同时也为中兽药制剂中常见非特定添加药物及残留的快速判定提供了可靠的方法。
1 材料与方法
1.1 仪器 UPLC Acquity -XEVO TQD 超高效液相色谱串联质谱仪, Masslynx色谱工作站(美国Waters 公司)、BEH C18色谱柱(2.1 mm×100 mm×1.7 μm)、CPA225D电子分析天平(德国Sartorius公司)。
1.2 试药与试剂 磺胺嘧啶对照品(批号:H0361504,中国兽医药品监察所,纯度99.5%),磺胺甲基嘧啶对照品(批号:G982705,Dr.Ehrenstorfer,纯度99.1%),磺胺二甲嘧啶对照品(批号:H0371406,中国兽医药品监察所,纯度99.2%),磺胺间甲氧嘧啶对照品(批号:C0031610,中国兽医药品监察所,纯度99.5%),磺胺甲噁唑对照品(批号:H0261106,中国兽医药品监察所,纯度99.8%),磺胺二甲氧嘧啶对照品(批号:C0061807,中国兽医药品监察所,纯度99.6%),磺胺喹噁啉对照品(批号:H0251407,中国兽医药品监察所,纯度99.6%),氧氟沙星对照品(批号:H0091210,中国兽医药品监察所,纯度99.5%),环丙沙星对照品(批号:130451-201904,中国食品药品检定研究院,纯度83.1%),恩诺沙星对照品(批号:H0081505,中国兽医药品监察所,纯度99.5%),多西环素对照品(批号:K0131507,中国兽医药品监察所,纯度85.2%),氟苯尼考对照品(批号:K0302010,中国兽医药品监察所,纯度99.6%),喹乙醇对照品(批号:C1572400,Dr.Ehrenstorfer,纯度97.8%),甲氧苄啶对照品(批号:100031-201606,中国食品药品检定研究院,纯度99.8%),金刚烷胺对照品(批号:G5410125,ANPEL,纯度97.5%),氯霉素对照品(批号:130555-201704,中国食品药品检定研究院,纯度99.8%),地塞米松对照品(批号:100129-201508,中国食品药品检定研究院,纯度99.8%),亲水亲油平衡材料(HLB SPE)固相萃取柱(200 mg,6 mL,华谱科仪(大连)科技有限公司)
供试品:麻杏石甘口服液(保定冀中药业有限公司,规格:100 mL/瓶,批号:202109029、202108028、202108027),乙腈(Merck公司,色谱纯),甲酸(Fisher Scientific,色谱纯,批号:106055A)。
1.3 液相色谱条件 以十八烷基硅烷键合硅胶为填充剂;乙腈(A)-0.1%甲酸(B)梯度洗脱流速:0.2 mL/min,柱温:30 ℃,进样量5 μL,梯度洗脱程序见表1,该液相色谱条件适用于1组的13种药物。
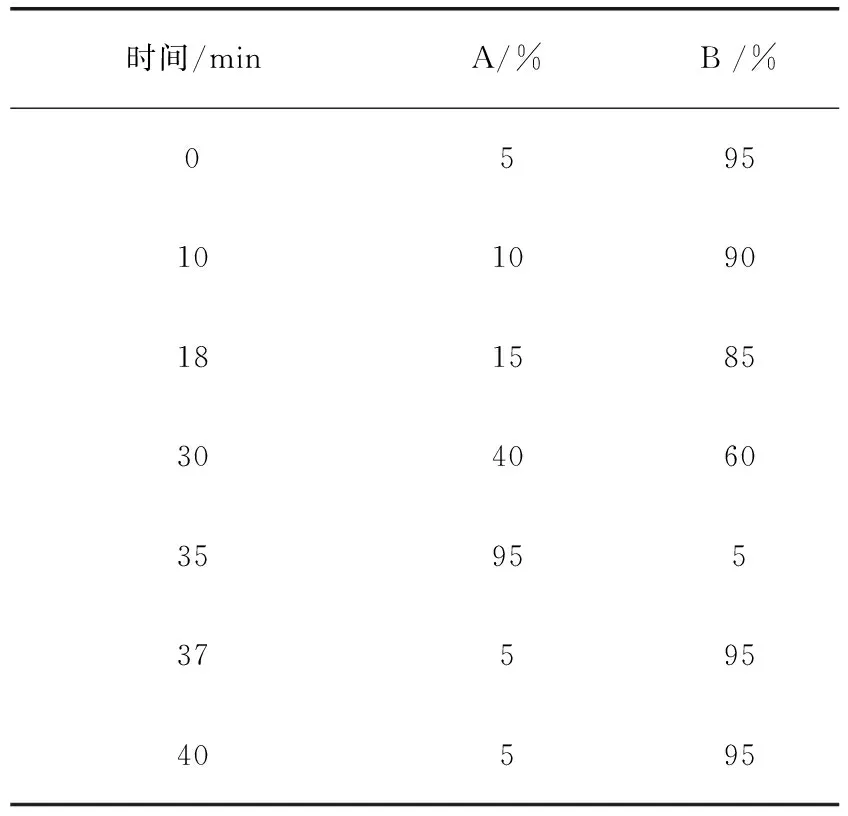
表1 梯度洗脱程序Tab 1 Program of gradient elution
以十八烷基硅烷键合硅胶为填充剂;乙腈(A)-0.1%甲酸(B)梯度洗脱流速:0.2 mL/min,柱温:30 ℃,进样量5 μL,梯度洗脱程序见表2,该液相色谱条件适用于2组的4种药物。
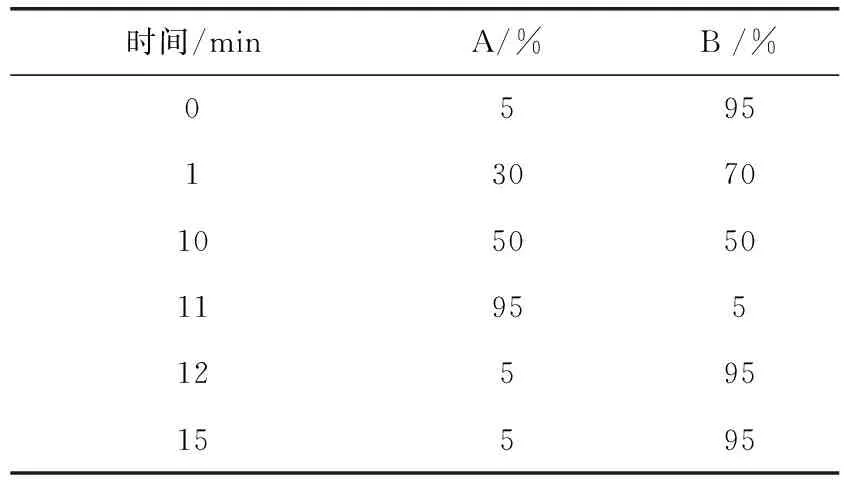
表2 梯度洗脱程序Tab 2 Program of gradient elution
1.4 质谱条件 电喷雾离子源,正离子扫描和负离子扫描;多反应监测模式;正离子模式毛细管电压3.0 kV,负离子模式毛细管电压2.0 kV;脱溶剂气温度500 ℃,脱溶剂气流速1000L/Hr;各化合物质谱采集参数见表3。
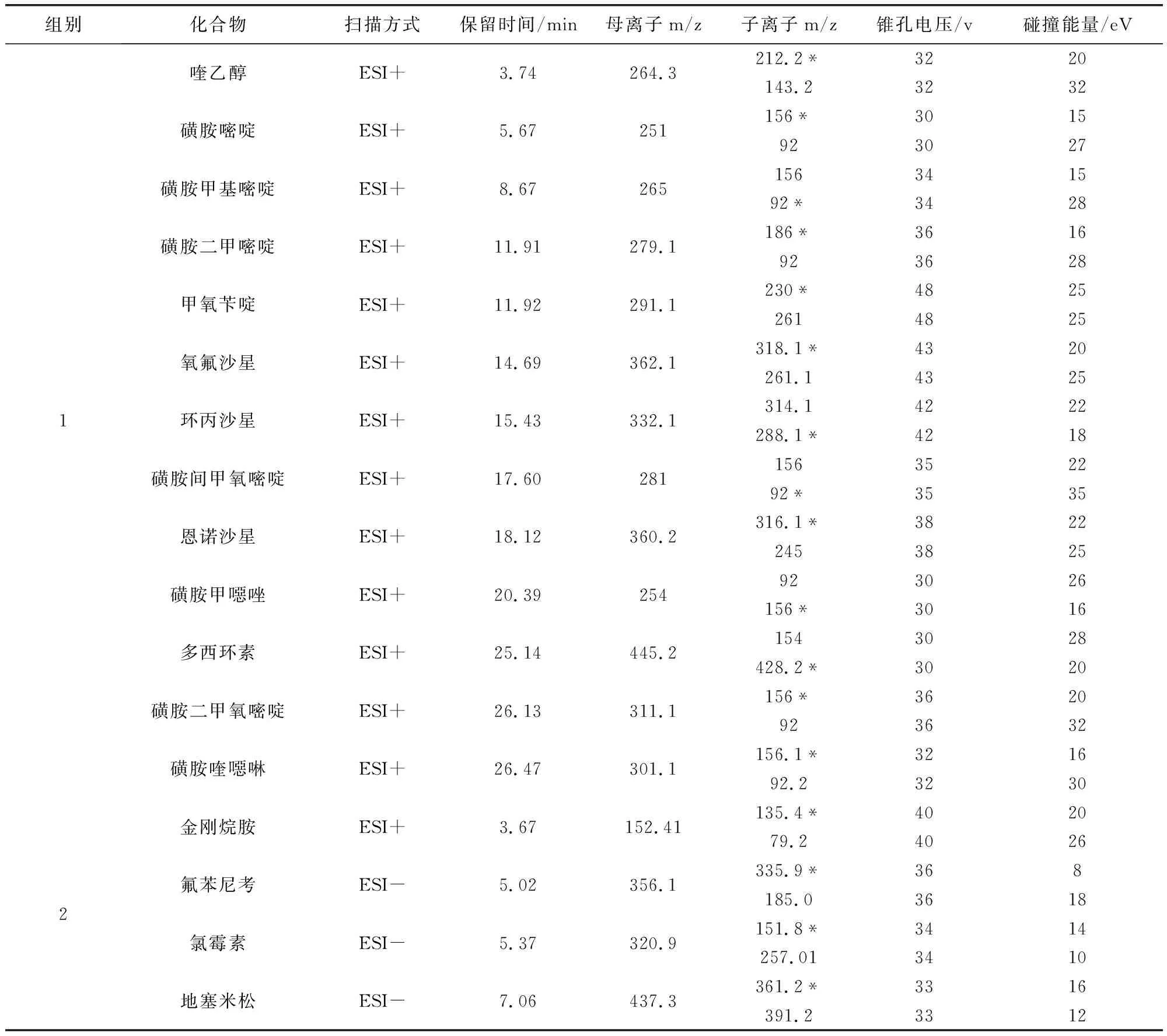
表3 17种化合物质谱参数Tab 3 Retention time and mainmass spectrametry parameters of 17 compounds
1.5 样品制备
1.5.1 阴性样品制备 麻杏石甘口服液,实验室自制。
1.5.2 单标标准储备液的制备 分别准确称取适量的各标准物质共计17种化合物,分别用甲醇溶解并定容,配制成 500 μg/mL 的单标标准储备溶液,于-20 ℃ 以 下避光冷冻保存。
1.5.3 混合标准储备液的制备 分别精密量取50 μL 17种化合物单标储备液于10 mL 容量瓶中,用甲醇溶解并定容至刻度, 配制成 2.5 μg/mL 的 17种化合物混合标准储备溶液,于-20 ℃ 以 下避光冷冻保存。
1.5.4 样品溶液的制备 精密量取2 mL麻杏石甘口服液于50 mL离心管中,加10 mL甲醇,涡旋2 min,超声提取5 min,5000 r/min离心5 min,取全部上清液通过HLB小柱,收集净化液,氮吹(45 ℃)至近干,加甲醇:0.1%甲酸=10:90定容至2 mL,摇匀,过滤,即得。
1.5.5 空白基质溶液的制备 精密量取阴性样品3 份,按照 1.5.4项下方法处理后即得空白基质溶液。
1.5.6 基质匹配标准储备溶液的制备 精密量取混合标准储备液50 μL置10 mL 容量瓶中,加空白基质溶液稀释至刻度,摇匀,滤过,即得 2.5 μg/mL 的基质匹配标准储备溶液。
1.5.7 阳性添加样品溶液的制备 精密量取混合标准储备液适量加入到阴性空白麻杏石甘口服液中,混匀,制成每1L分别添加10、25、50 μg(喹乙醇添加25、50、100 μg)的阳性添加样品,每个添加量制备3份平行样,按1.5.4所述方法制备阳性添加样品,进行测定,每份样品至少进样2次,取平均值。
1.5.8 基质标准曲线的制备 准确吸取1 mL基质匹配标准储备溶液(2.5 μg/mL),至10 mL量瓶中,用空白基质溶液稀释至刻度,摇匀,得0.25 μg/mL的基质标准溶液。分别精密吸取该基质标准溶液适量,用空白基质溶液稀释制成5、10、25、50、100、120 ng/mL的标准物质系列工作液。
1.5.9 检出限和定量限 在空白基质溶液中添加适量的17种化合物的基质匹配标准储备溶液,按照1.5.4所述方法制备样品,照1.4所述仪器条件进行测定,进行检出限和定量限的测定。
3 结果与分析
3.1 线性范围 按 1.5.8方法配制的 17种化合物基质混合标准工作曲线在选定的仪器条件下测定。17种化合物基质混合标准溶液总离子流图见图1~2,各化合物分离度良好,响应灵敏。以各化合物定量离子色谱峰面积 A 为纵坐标,各化合物质量浓度(ng/mL)为横坐标,绘制线性回归方程。结果表明:17种化合物在 10~100 ng/mL(喹乙醇25~120 ng/mL)浓度范围内线性关系良好,相关系数r均大于 0.99,17种化合物的线性回归方程及相关系数见表4。

图1 第1组13种化合物基质标准溶液总离子流图Fig1 Total ion flow chromatograms of 13 compounds matrix standard solution in the first group
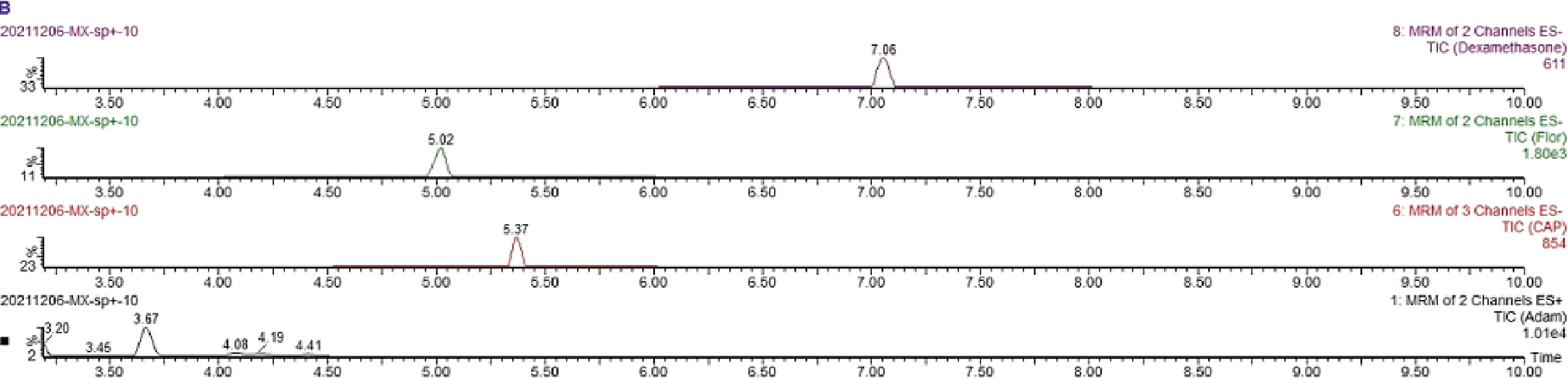
图2 第2组4种化合物基质标准溶液总离子流图Fig2 Total ion flow chromatograms of 4 compounds matrix standard solution in the second group
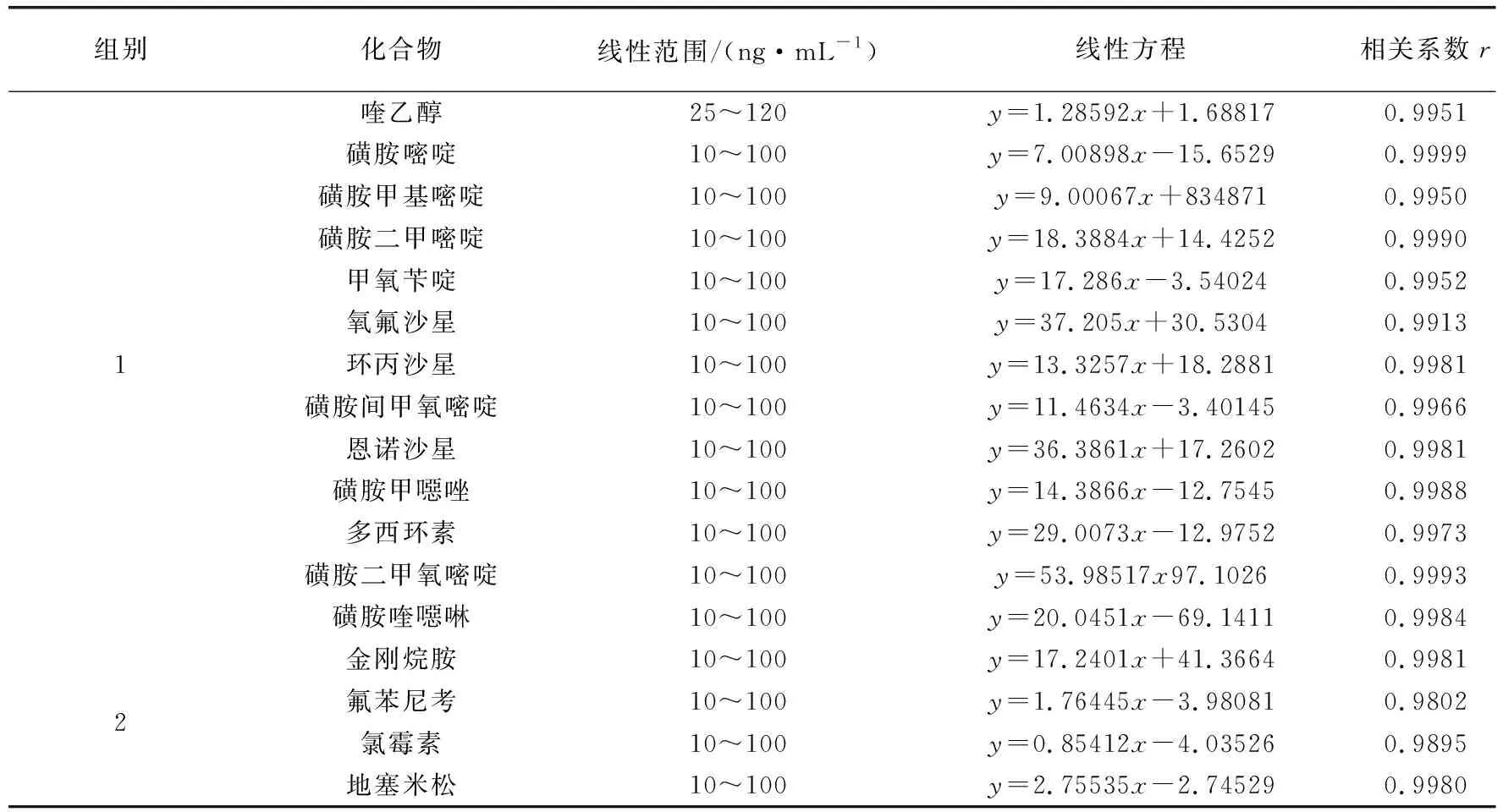
表4 17种化合物线性范围、线性方程、相关系数Tab 4 Mainmass spectrametry parameters of 17 compounds
3.2 精密度考察 精密吸取基质匹配标准溶液 (25 ng/mL)5 μL,连续进样 6次。以峰面积计算,得出17种标准物质的RSD为1.9%,表明仪器的精密度良好,符合要求。
3.3 检出限和定量限 测定结果表明:在相应的保留时间,空白样品对 17 种目标化合物无干扰,空白样品溶液总离子流图见图3~图4。依据特征离子质量色谱峰信噪比S/N≥3的浓度为方法检出限,其中仅喹乙醇的检出限为10 ng/mL,剩余16种药物的检出限均为5 ng/mL,特征离子质量色谱峰信噪比S/N≥10的浓度为方法定量限,其中仅喹乙醇的定量限为25 ng/mL,剩余16种药物的定量限为10 ng/mL。
图3 第1组空白样品溶液总离子流图Fig3 Total ion flow chromatograms of blank samples in the first group

图4 第2组空白样品溶液总离子流图Fig4 Total ion flow chromatograms of blank samples in the second group
3.4 准确度 分别制备10、25、50 ng/mL三个添加浓度(喹乙醇添加浓度为25、50、100 ng/mL),经测定,平均回收率在73.87%~116.43%之间,RSD为1.25~8.44%(表5)。
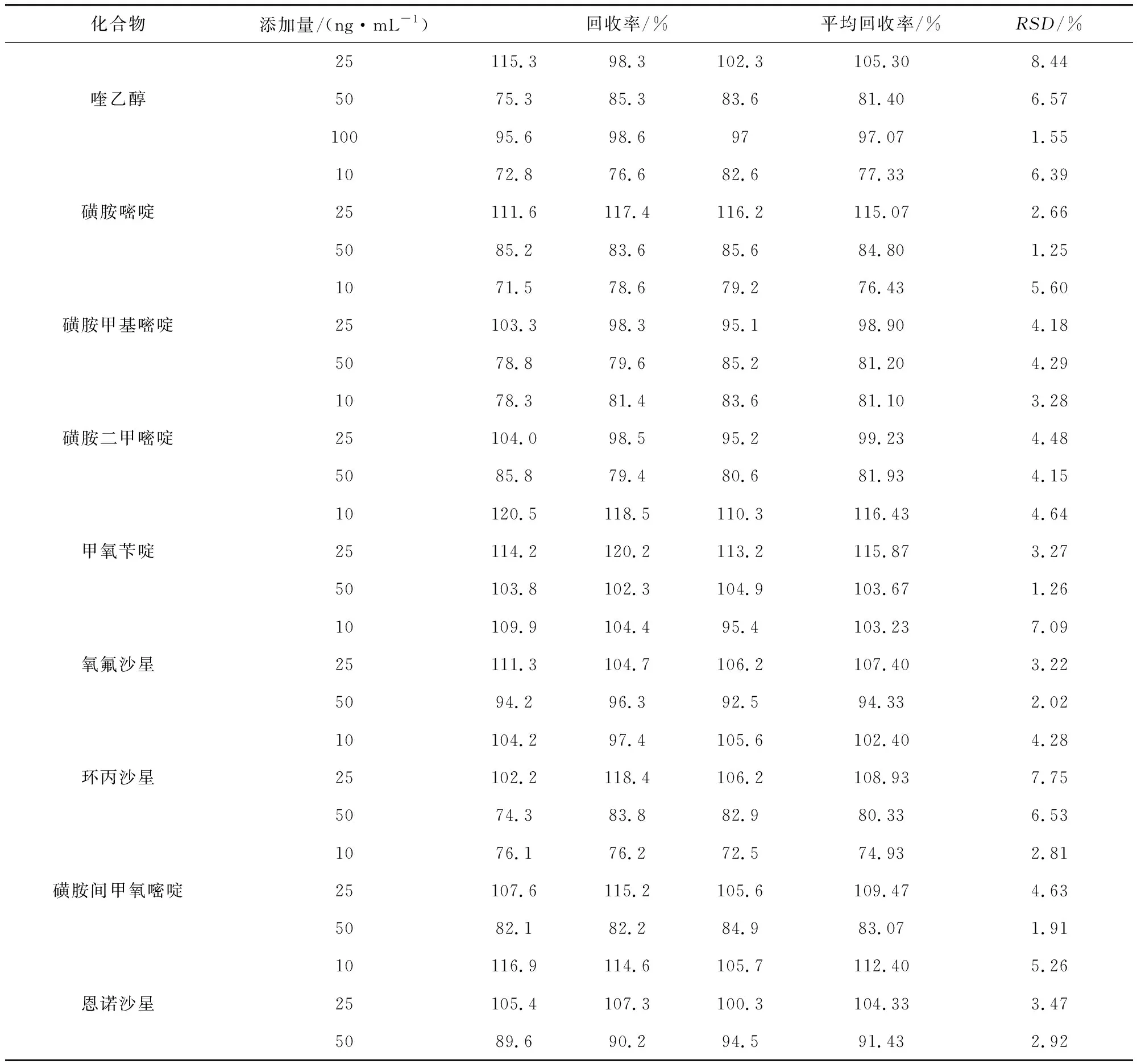
表5 回收率试验结果Tab 5 Results of recoveries
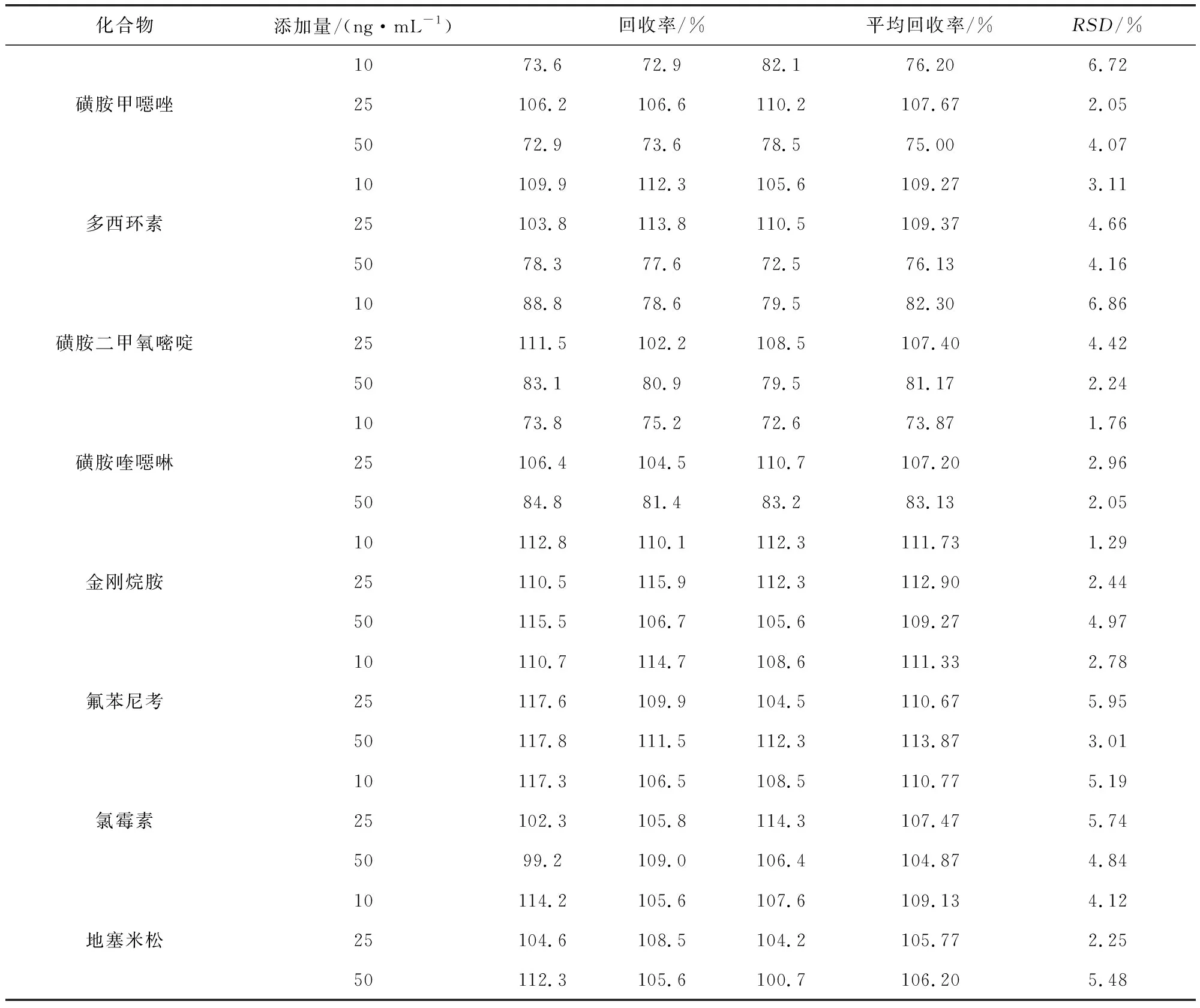
续表5
4 讨论与结论
4.1 筛查物质种类确定 据文献[13-14]报道,市售中兽药注射液、口服液、散剂中非法添加药物有氯霉素(酰胺醇类药物)、氟喹诺酮类药物、抗病毒药物(金刚烷胺)、喹诺酮类药物,在与各养殖集团合作过程中,发现养殖企业对上述品类中17种药物的关注度较高,因此,本文中就17种药物的检测开展研究。
4.2 净化条件的选择 在前期的试验过程中,我们曾参照农业农村部公告289号采用稀释法(甲醇、乙腈)进行供试品前处理的制备,具体操作方法为取供试品1 mL,加甲醇4 mL,涡旋混匀后,离心,取上清1 mL,加0.1%甲酸溶液1 mL,混匀,过0.22 μm滤膜,上机检测。但是长时间运行后供试品溶液中未去除的复杂基质会累积在锥孔处,有大量的黑褐色斑块出现,需要经常性拆卸锥孔进行清理维护,采用与本文相同的流动相体系,喹乙醇的检出限为50 ng/mL,定量限为100 ng/mL,剩余16种药物的检出限为25 ng/mL,定量限为50 ng/mL,难以满足养殖集团的要求。在此基础上尝试采用甲醇提取继而采用透过法通过亲水亲油平衡材料(HLB SPE)固相萃取柱净化,可使供试品溶液的颜色由红棕色改为浅黄色,大大降低基质干扰程度,满足检测需求。
4.3 改进计划 经过比对,对畜禽养殖过程中常用到的双黄连口服液、清瘟解毒口服液、清解合剂和板青颗粒均进行了相关的研究,对于基质的干扰排除分别采用了通过亲水亲油平衡材料(HLB SPE)固相萃取柱、QuEChers净化管、GCB固相萃取小柱等前处理方法,不同产品中物质的响应受到不同程度的干扰,后期还需进行详细的研究,以期能够建立针对各产品的快速、高效、准确的方法便于进行安全性的质量控制。
4.4 结论 研究建立了一种系统筛查中兽药口服液制剂中17种化合物的检测方法,发挥了液相色谱串联质谱准确灵敏的优势,首次建立了中兽药液体制剂麻杏石甘口服液中多种非特定非法添加/残留物质的检测方法,有效加强了兽药生产企业对于产品质量的控制,建立了中兽药制剂中动物源性食品禁限用兽药的检测、判定的科学依据,增强了动物源性食品的安全屏障。该方法操作简单,净化效果好,准确度、灵敏度高,检测限低,适用于麻杏石甘口服液的安全性检测。
