密闭熔融-离子色谱法测定土壤中氟化物
2022-06-28毕建玲徐标孙鹏飞高玉花王飞飞邵长伟
毕建玲,徐标,孙鹏飞,高玉花,王飞飞,邵长伟
(1.山东省物化探勘查院,济南 250013; 2.山东省生态环境监测中心,济南 250101)
氟是自然环境中广泛分布且与人体健康密切相关的微量化学元素之一[1],土壤中的氟主要来源于土壤母质、自然环境以及工农业生产排放的含氟气体等。土壤中氟的含量在很大程度上取决于土壤中无机组分的矿物组成[2],而土壤环境中的氟是水和食物链中氟的主要来源[3-4]。研究发现,氟是人体一切矿化组织的重要成分,食物或饮水中氟缺乏,会影响人体牙齿和骨骼组织代谢,而氟的过量摄入则会造成人体氟中毒[5-6],主要临床表现为釉斑牙,严重者可导致氟骨病。随着工农业的发展,各种污染物排放到土壤中,土壤中的氟会迁移至地表水和地下水中,造成水源型氟中毒。氟在植物体内的累积随着植物种类不同而有所差异,在植物体内一些无机氟化物甚至可以转化为毒性更强的有机氟化物。氟在动植物体内无生物降解作用,严重污染时会直接危害人类和动植物,即使低水平的污染也能通过生物富集和食物链作用对人体和动植物造成一定的危害[7-9]。土壤氟化物污染日趋严重,已经引起了人们的广泛关注,因此及时了解土壤动态,快速准确分析土壤中氟化物具有重要现实意义。
目前氟化物的测定方法主要有离子选择电极法[10-13]、分光光度法[14-16]、气相色谱法[17-19]和离子色谱法[20-23]。离子选择电极法自动化程度低,稳定性较差,检出限较高;分光光度法操作步骤繁琐,过程复杂,耗时长;气相色谱法样品处理步骤繁琐,使用有机试剂对环境不友好。离子色谱法可用于多种无机阴离子和有机酸的同时分析,具有操作简单、多组分同时测定、成本低等优点,较适合痕量分析,而且离子色谱法实现了自动进样,对于大批量样品的测定还可节省大量试剂。陈静等[20]采用IonPac AS11-HC 型阴离子分析柱(250 mm×4.0 mm),用30 mmol/L 的KOH 溶液作为淋洗液,测定土壤中的有效氟;赵怀颖等[21]采用高温热解法处理样品,利用Dionex lonPac AS18 阴离子分析柱(4 mm×250 mm),以20 mmol/L 的NaOH 溶液作为淋洗液,测定植物中的氟,均取得良好的效果。酸溶条件下的低温增压溶矿技术比较成熟,但低温密闭碱熔技术在国内分析测试行业缺少实验与应用,采用低温密闭碱熔-离子色谱法测定土壤中的氟化物亦未见报道。笔者结合实际工作,研究了密闭熔融法样品处理条件,优化了离子色谱仪的工作参数,建立了测定土壤中氟化物的密闭熔融-离子色谱方法,并取得良好效果。
1 实验部分
1.1 主要仪器与试剂
离子色谱仪:ICS-1100 型,配有IonPac AS19 型色谱柱(250 mm×4.0 mm)、ADRS 600-4 mm 型连续自动再生微膜抑制器、电导检测器,美国赛默飞世尔科技公司。
台式电热鼓风干燥箱:101 型,北京永光明医疗仪器厂。
密闭防腐熔样罐:JKHF-300 型,青岛济科实验仪器有限公司。
电子天平:BSA124S 型,感量为0.000 1 g,赛多利斯科学仪器(北京)有限公司。
氟离子标准溶液:1 000 mg/L,GSB 04-1771-2004,国家有色金属及电子材料分析测试中心。
氢氧化钠:优级纯,国药集团化学试剂有限公司。
硫酸溶液:2 mol/L,移取42 mL 硫酸缓慢加至700 mL 水中,搅拌均匀。
阳离子交换树脂:732 型,用水浸泡阳离子交换树脂,清洗数遍后,将树脂装入直径约1.5 cm、长约30 cm 的玻璃柱中,顶端与梨形分液漏斗衔接。于分液漏斗中加入2 mol/L硫酸溶液150 mL,以约1.5 mL/min 流量流经交换柱。用水以同样流量流经交换柱,直至流出液洗至无硫酸根。再生的树脂以真空抽滤至干,装瓶备用。
土壤标准物质:编号分别为GBW 07388、GBW 07390、GBW 07562、GBW 07573、GBW 07446、GBW 07407,中国地质科学院地球物理地球化学勘查研究所。
土壤样品:某农用地土壤。
实验用水为去离子水。
1.2 溶液配制
氟离子标准使用溶液:100 mg/L,吸取氟离子标准溶液10 mL 至100 mL 容量瓶中,用去离子水稀释至标线,摇匀。
系列氟离子标准工作溶液:分别吸取氟离子标准使用溶液0、1.0、2.0、5.0、7.0、10.0 mL于6只100 mL 容量瓶中,用水稀释至标线,摇匀,配制成氟离子质量浓度分别为0、1.0、2.0、5.0、7.0、10.0 mg/L的系列标准工作溶液。
1.3 样品处理
准确称取0.100 0 g 样品,置于30 mL 防腐熔样罐内,加入0.800 g 氢氧化钠熔剂,加入1 mL 去离子水,密闭内胆,放入烘箱内,于220 ℃加热熔融6 h,取出充分冷却后,打开内胆,用去离子水提取样品,提取液转移至25 mL 具塞比色管中,用去离子水定容至标线,以3 000 r/min 离心后,吸取20 mL上清液置于100 mL 烧杯中,加入732 型阳离子交换树脂5 g,在静态交换过程中摇动2~3 次,直至溶液呈微酸性后再静置30 min,移取上清液过0.22 μm 滤膜,待测。
1.4 仪器工作条件
色谱柱:Dionex IonPac AS19柱(250×4.0 mm,美国赛默飞世尔科技公司);保护柱:Dionex IonPac AS19 柱(50×4.0 mm,美国赛默飞世尔科技公司);抑制器:连续自动再生膜阴离子抑制器,电流为50 mA;检测器:电导检测器;淋洗液:20 mmol/L的KOH 溶液,流量为1.00 mL/min;进样体积:25 μL;电导池温度:35 ℃;柱温:30 ℃。
2 结果与讨论
2.1 氢氧化钠熔剂用量选择
氟化物的反应活性与其它卤化物有显著不同,由于其半径/电荷比小,熔剂化倾向更强,更趋近于氢氧化物,因此熔剂一般选择氢氧化钠或氢氧化钾。氢氧化钠熔点为318 ℃,氢氧化钾熔点为380 ℃,理论上都能很好地分解熔融土壤样品,但考虑到熔剂的成本,一般不选用氢氧化钾,故选用氢氧化钠作为熔剂。
样品与熔剂质量比是熔融的重要条件,合适的比例既能保证样品完全熔融,又能节省熔剂并减少熔剂带来的离子干扰。在前人研究的基础上,选择样品与熔剂质量比分别为1∶5、1∶6、1∶8、1∶10进行试验。分别称取国家标准物质GBW 07388、GBW 07390、GBW 07562,各0.100 0 g,每种标准物质分别加入0.5、0.6、0.8、1.0 g氢氧化钠,然后各加入1.0 mL 去离子水,于220 ℃下熔融6 h,在1.4 仪器工作条件下平行测定3 次,以3 次测定结果的平均值作为测定值,结果如图1 所示。由图1 可以看出,当样品与熔剂质量比为1∶5 时,3 种标准物质的回收率均在80%~85%范围内,相对较低,说明氢氧化钠用量不足,不能完全熔融氟化物。随着氢氧化钠用量增加,回收率逐渐升高,当样品与氢氧化钠质量比达到1∶8 时,3 种标准物质的回收率均达到100%左右,并趋于稳定,因此选择样品与熔剂质量比为1∶8。
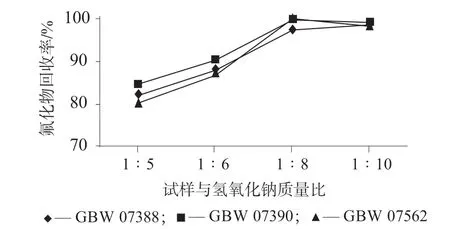
图1 不同氢氧化钠用量时氟化物的回收率
2.2 熔融温度选择
温度是样品熔融的关键因素之一。为了确定土壤熔融的最佳温度,选取3 种国家标准物质GBW 07388、GBW 07390、GBW 07562,分别平行称取3份,各0.100 0 g,置于密闭熔样罐中,固定其它试验条件不变,分别考察熔融温度为180、200、220、240℃时对样品熔融效果的影响,结果如图2 所示。由图2 可以看出,随着熔融温度的升高,3 种标准物质的回收率逐渐升高,当熔融温度为220 ℃时,3 种标准物质的回收率均接近100%,并趋于稳定,可以确定样品完全熔融,故选择样品熔融温度为220 ℃。
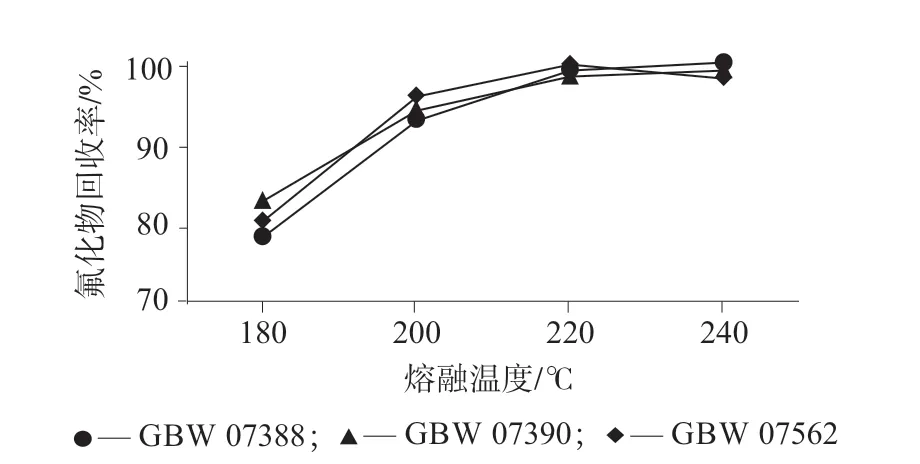
图2 不同熔融温度时氟化物的回收率
2.3 熔融时间选择
为确定土壤中氟化物熔融的最佳时间,选取3种国家标准物质GBW 07388、GBW 07390、GBW 07562,分别平行称取3 份,各0.100 0 g,置于密闭熔样罐中,加入氢氧化钠熔剂0.800 g,设置熔融温度为220 ℃,分别设置熔融时间为2、4、6、8、10 h,在1.4 仪器工作条件下进行测定,结果如图3 所示。由图3 可以看出,3 种标准物质在熔融时间达到6 h 以后,氟化物的回收率均达到100%左右,并趋于平稳,可以确定氟化物完全熔融,故选择熔融时间为6 h。
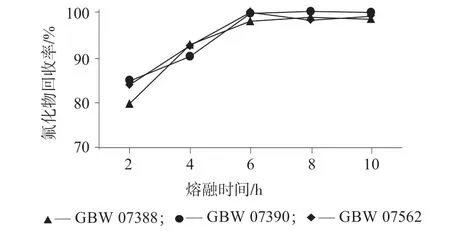
图3 不同熔融时间时氟化物的回收率
2.4 样品溶液预处理
碱熔后的样品含有大约0.80 mol/L氢氧化钠溶液,不能直接进入离子色谱分析,因为碱含量过高会造成色谱柱污染,水系统峰消失而出现一个正的碱峰;另外高浓度氢氧化钠溶液将起淋洗液的作用,使色谱峰保留时间发生改变,导致各组分的出峰时间前移,致使氟的色谱峰与碱峰重叠,而无法准确测定氟的含量。试验采用732 型阳离子交换树脂对样品溶液进行预处理,732 型强酸性阳离子树脂含有大量的强酸性基团,如磺酸基(—SO3H),容易在溶液中离解出H+,故呈强酸性,树脂离解后,本体所含的负电基团,如—SO3,能吸附结合溶液中的其它阳离子,使树脂中的H+与溶液中的阳离子互相交换。强酸性树脂的离解能力很强,在酸性或碱性溶液中均能离解和产生离子交换作用。将25 mL 样品溶液置于100 mL 烧杯中,加入5 g 阳离子交换树脂,在静态交换过程中需摇动2~3 次,直至溶液呈微酸性后再静置30 min。移取上清液经0.22 μm 滤膜,在1.4 仪器工作条件下进行测定。结果表明,经732阳离子交换树脂处理后,消除了碱峰影响,基线平稳,分离效果较好。
2.5 淋洗液浓度选择
在获得较好分离结果的前提下,分析时间越短越好,但淋洗液的浓度增大会增大背景电导,导致测定灵敏度降低,降低弱保留氟离子的分离度。选择实际土壤样品,按照1.3 样品处理方法,平行处理3份样品溶液,以氢氧化钾溶液为淋洗液,考察氢氧化钾溶液浓度分别为15、20、25、30 mmol/L 时的分离效果。结果显示,氢氧化钾溶液浓度越高,各组分的保留时间越短,分离效果越差,当氢氧化钾溶液浓度达到30 mmol/L 以上时,各组分出峰(F-、Cl-、NO2-、Br-、NO3-)相互干扰严重;当氢氧化钾溶液浓度为25 mmol/L 时,对各组分出峰稍有干扰;当氢氧化钾溶液浓度为15、20 mmol/L 时,各组分分离效果较好,但氢氧化钾浓度为15 mmol/L 时保留时间较长,综合考虑,选择氢氧化钾溶液浓度为20 mmol/L。
2.6 淋洗液流量选择
淋洗液流量影响色谱柱和系统的压力、离子的分离度以及洗脱时间,特别是对标准曲线法定量的分析结果影响较大,一定范围内可通过增加流量来缩短保留时间而并不明显降低分离效率。选用实际土壤样品,按照1.3 样品处理方法,平行处理3 份样品溶液,以20 mmol/L 氢氧化钾溶液为淋洗液,流量分别设定为0.8、0.9、1.0、1.2 mL/min,考察不同流量下氟离子的色谱响应,试验数据列于表1。由表1可知,淋洗液流量越大,保留时间越短,但柱压随着流量的增大而上升,同时随流量增大色谱峰高和色谱峰面积均呈下降趋势,但淋洗液流量过大会降低F-和C1-的分离度。综合考虑保留时间和分离度,选择淋洗液流量为1.0 mL/min。
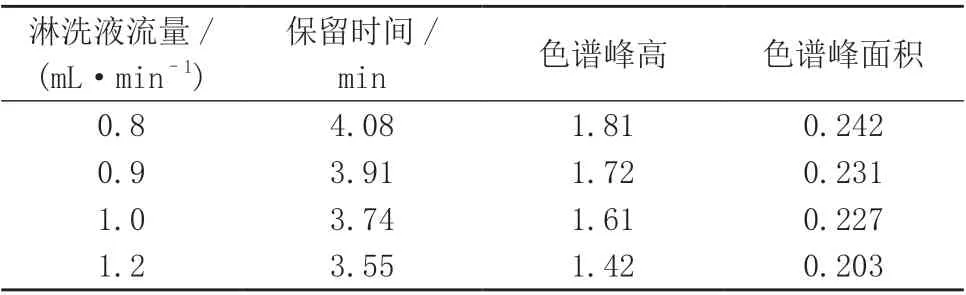
表1 不同淋洗液流量下F-的色谱响应值
2.7 线性方程与检出限
在1.4 仪器工作条件下,对系列氟离子标准工作溶液进行测定,以氟离子质量浓度为横坐标,以色谱峰面积为纵坐标,绘制标准工作曲线,计算得线性方程为y=0.429x,相关系数为0.999 8,氟离子质量浓度线性范围为0~10 mg/L。
在1.4 仪器工作条件下,对空白溶液重复测定12 次,以3 倍标准偏差对应的质量浓度作为方法检出限,根据称样质量和定容体积换算成样品中的含量,以质量分数表示,得方法检出限为2.5 mg/kg。
2.8 精密度试验
选择3 份土壤样品(编号分别为1#、2#、3#),按1.3 样品处理方法分别平行处理7 份样品溶液,在1.4 仪器工作条件下进行测定,计算测定结果的相对标准偏差,结果列于表2。由表2 可知,测定结果的相对标准偏差为1.49%~5.53%,表明该方法精密度良好,满足要求[25]。

表2 精密度试验结果
2.9 准确度试验
选取3 种标准物质GBW 07446、GBW 07388和GBW 07407,按1.3 样品处理方法分别平行处理6 份样品溶液,在1.4 器工作条件下进行测定,计算测定平均值与标准值的相对误差,结果见表3。由表3 可知,测定结果均在标准值不确定度范围内,相对误差为1.09%~1.83%,表明该方法准确度满足测定要求[25]。
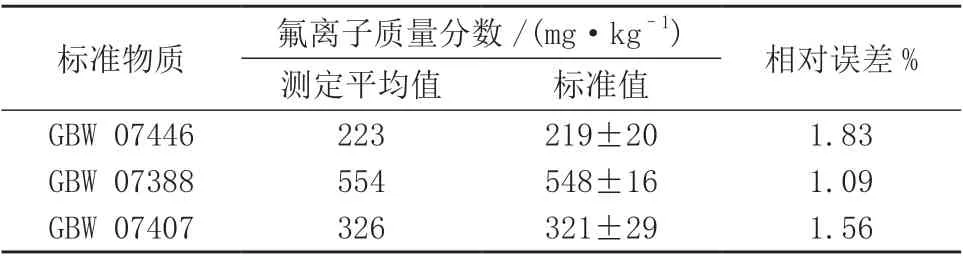
表3 准确度试验结果(n=6)
2.10 比对试验
选取6 种土壤标准物质GBW 07573、GBW 07446、GBW 07407、GBW 07388、GBW 07390 和GBW 07562,按1.3 样品处理方法各平行处理3 份样品溶液,分别采用高温碱熔-离子选择电极法[24]和密闭熔融-离子色谱法进行测定,结果见表4。由表4 可知,两种方法的测定结果相近。经t检验,两种方法的测定结果无显著性差异(t测<4.30)。离子选择电极法虽然操作简便,但每个样品测试稳定时间较长,测定后需用纯水将电极反复冲洗干净,且需要人工操作;离子色谱法操作简便,每个样品进样后自动测定,而且测定大批量样品时试剂用量少,降低了环境污染。
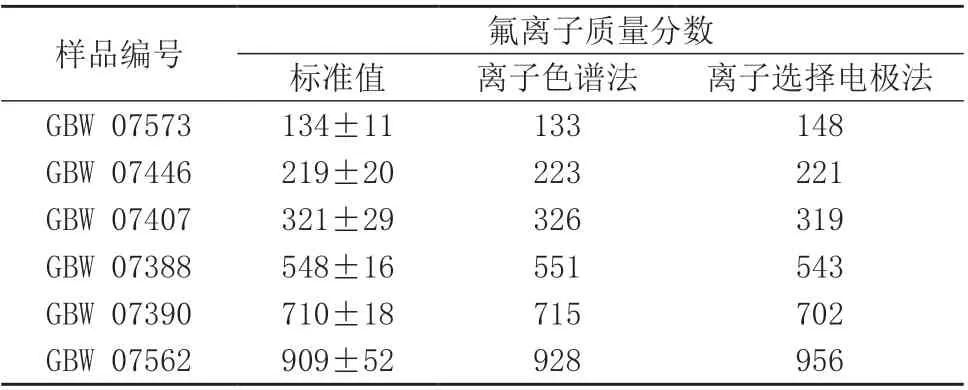
表4 离子选择电极法和离子色谱法测定结果 mg/kg
3 结语
建立了测定土壤中氟化物的密闭熔融-离子色谱法。样品在220 ℃下经氢氧化钠密闭熔融6 h,可以将土壤中的氟化物完全溶解,溶解液经732 型强酸性阳离子交换树脂处理,采用Ion Pac AS19 色谱柱,以20 mmoL/L 氢氧化钾溶液为淋洗液测定土壤中的氟化物。该方法精密度好,准确度高,检出限低,操作简便快速,劳动强度低,对环境无污染,可以满足实际样品的分析要求。
