抗体药物的药物代谢动力学特点及研究进展
2022-06-28张金桂王超
张金桂,王超
作者单位:海南省人民医院,aI期临床试验病房,b药学部临床药学室,海南 海口 570311
抗体是由效应B细胞产生的一种具有免疫功能的球蛋白。自学者Kohler 和Milstein 在1975 年取得了杂交瘤细胞技术制备单克隆抗体(mAb)的突破性进展以来,针对恶性肿瘤、自身免疫疾病等研发的抗体药物不断发展[1]。截至2020 年,全球范围内销售金额最高的10 种药物中有4 种是抗体药物,其总销售金额高达350亿美元[2-3]。
抗体药物包括mAb和基于经典抗体结构衍生的具有类似结构或功能的新型抗体药物。抗体药物由于结构、相对分子质量和亲水性等特征而具有不同的代谢机制,研究药物代谢动力学特征有利于制定更加合理的临床给药方案,优化抗体分子结构设计。本研究分别对抗体药物的基本结构、抗体药物的药物代谢动力学(PK)特征进行综述,并对新型抗体药物的PK研究进展进行介绍,为抗体药物的应用提供参考。
1 抗体药物结构
经典IgG 型抗体结构见图1,骨架可分为抗原结合区(Fab 结构域)和恒定区(Fc 结构域)两部分。Fab 区主要与抗原表位结合,Fc 区与细胞表面特异性Fc 受体结合,介导抗体的效应子功能、体内循环和转运等功能[4]。
抗体分子是一种糖蛋白,糖链通常修饰于抗体分子的CH2亚结构域上,也有少数修饰于抗体其他亚结构域中[5],例如西妥昔单抗(Cetuximab)[6]在CH2与VH亚结构域均带有糖基化修饰。抗体的糖链一般为具有2~3 个分支的触角型结构,由N-乙酰葡萄糖胺(N-acetylglucosamine)、岩藻糖(Fucose)、甘露糖(Mannose)、半乳糖(Galactose)、唾液酸(Sialic Acid)等单糖连接组成[5]。糖链以N-糖苷键连接在抗体分子的保守糖基化位点上,即连接在天冬酰胺-X-丝氨酸/苏氨酸(其中X 为除脯氨酸以外的任意氨基酸)序列中的天冬氨酸残基上。
通过改造mAb 经典结构可以得到多种结构不同并发挥特定功能的新型抗体药物:包括纳米抗体(Nb)、双特异性抗体(BsAb)、抗体药物偶联物(ADC)等。见图2。
Nb是摒弃了经典mAb的一部分恒定区,仅将可变区VH和VL结构域经由特定的方式均价连接而成的单链可变片段(scFv)或仅保留单一VHH 结构域(骆驼科重链抗体的抗原结合区)如卡普赛珠单抗(Ca‑placizumab-yhdp)。部分Nb 在分子设计时也会考虑将Fc片段与scFv或VHH融合。Nb分子量远小于经典mAb,一般仅有15~100 kDa[7-8]。
BsAb 是将两个或多个抗原结合区整合到同一抗体分子上构建而成的新型分子,以多种抗原作为同一靶点的抗体药物,如博纳吐单抗(Blinatumom‑ab)。目前共有60 多种不同的BsAb 分子结构,不同BsAb 分子其模块结构、分子大小,预期药物代谢动力学行为均存在差异[9]。
ADC 是通过特定化学接头将细胞毒性药物与抗体分子进行偶联的新型分子,曲妥珠单抗-美坦新偶联物(Ado-trastuzumabemtansine)。常规ADC分子中药物-抗体摩尔比(DAR)为3.5~4.0[10]。
2 抗体药物的吸收
不同于小分子化学药物可以口服后经由胃肠道吸收,抗体药物作为大分子会在胃肠道内被消化降解。目前,全球范围内尚未有经口服给药的抗体药物获批上市。大多数抗体药物均采用静脉注射给药途径,不涉及吸收过程。少数需要长期给药的抗体药物采用皮下注射或肌肉内注射途径给药,如依那西普(Etanercept)[11]和帕丽珠单抗(Palivizumab)[12]。一般认为通过此类血管外途径给药的抗体药物是通过多孔淋巴系统吸收并缓慢进入血管,整个过程持续数小时甚至数天,因此经皮下注射或肌肉内注射途径给药的抗体血药浓度达峰时间也相应变长[13]。虽然皮下或肌内注射给药不可避免地存在一定程度的抗体降解,但生物利用度依然可以达到50%~100%[14]。
3 抗体药物的分布
血液中的抗体药物主要通过对流(Convection)方式转运至组织,受到缓慢对流摄取和抗体从组织快速清除的共同作用,组织抗体浓度一般低于血液抗体浓度。在高度灌注且脉管系统通透性较高的组织(如骨髓、脾脏和肝脏)中抗体分布会较高[15]。由于抗体药物具有靶向性,虽然其表观分布容积较低,但靶组织、靶器官的药物分布仍高于其他组织。靶向能力不同的抗体药物其体内分布情况大相径庭。针对抗体药物体内分布的研究与讨论多集中在靶向分布情况上,并着重关注于靶组织、靶器官内的具体分布行为[16]。Khaowroongrueng 等[17]利用大孔微透析技术进行抗体药物R7072的肿瘤间质分布测定时发现该抗体在给药后的肿瘤间质浓度与血清浓度之比呈现剂量依赖性的增加,即说明了R7072显示出良好的靶向分布性质。
4 抗体药物的代谢
不同于传统小分子药物在肝脏中由细胞色素P450 酶介导的经典代谢过程,抗体药物不能经由肝脏或肾脏直接代谢。抗体药物具有多种独特的代谢方式,如与靶点介导的药物处置(TMDD)、新生儿Fc受体(FcRn)介导的循环再利用、糖基化修饰和电荷异质性对清除速率的影响以及与抗药物抗体相结合导致清除加快。这些代谢方式使得抗体药物在体内的代谢行为十分复杂,针对其设计更安全长效的抗体药物也是近年来生物药物研发领域的热点。
4.1 TMDDTMDD 是一种非线性的药物代谢方式,即药物与作用靶点(如酶、受体、转运蛋白等)结合并发挥药理作用后导致的剂量依赖性代谢趋势变化。
抗体药物与靶点抗原结合后,抗原-抗体复合物被靶细胞内化而降解。当靶组织内抗体药物的量远小于靶点的量或二者亲和力不足时,靶点未被抗体结合饱和,此时TMDD 效应是抗体药物清除的主要因素。随着抗体药物浓度的增加,靶点逐渐被结合抗体饱和,此时TMDD效应也趋于饱和,抗体的清除率随之急剧下降。在抗体药物浓度足够高时非线性途径的TMDD效应对其代谢的影响可以忽略不计,抗体在体内清除过程接近于一级动力学消除[16]。与小分子药物相比,TMDD 效应在抗体药物中表现得更为显著[18]。在实际临床前研究中,为了确定抗体药物是否存在TMDD 效应,可通过单次递增剂量的体内PK 研究,将抗体药物清除率或半衰期对给药剂量作图分析获得结果。
尽管早期研究普遍认为TMDD根据其发生机制主要影响靶点为细胞膜表面抗原的药物,但近年来一些研究表明,靶点为可溶性抗原的抗体药物代谢也会存在TMDD效应。Brenner等[19]研究了一种靶向结缔组织生长因子(CTGF)的人源mAb FG-3019,该抗体被证实存在TMDD效应导致的快速体内清除。
4.2 FcRn 介导的循环再利用FcRn 是在多种细胞中表达的特定受体蛋白,可与IgG 型抗体的Fc 区结合并参与抗体分子的转运[20]。FcRn 通过高度pH依赖的细胞再循环机制避免了抗体在细胞内的降解并将抗体转运至胞外,使得抗体药物能够维持一定的血药浓度并具有较长半衰期。
FcRn 与抗体Fc 区的相互作用发生在抗体的CH2和CH3亚结构域界面,二者相互作用高度依赖于环境pH,抗体在酸性环境下(pH=5.5~6.5)以高亲和力结合FcRn,但随着pH 升高至中性(pH=7.4),抗体与FcRn结合的亲和力会大幅下降。因此,被胞饮作用摄入细胞的抗体在酸性核内体中与FcRn结合,被FcRn 转运回到细胞表面并在胞外中性生理pH 下与FcRn 解离,抗体被释放并再次进入循环,未与FcRn结合的抗体或pH 依赖程度不高的抗体会被胞饮进溶酶体并被降解[20]。
通过蛋白质工程的手段增强抗体与FcRn 在酸性pH下的结合,同时降低抗体与FcRn在中性pH下的结合,使抗体药物与FcRn结合规律最有利于实现由FcRn介导的抗体循环再利用,延长抗体药物半衰期,成为近年来抗体药物分子设计的热点方向之一。Booth 等[21]设计了一组抗体药物,其pH 依赖性的FcRn 结合力提升约30 倍。PK 数据显示,此组抗体药物在转基因鼠体内的半衰期延长9 倍,在食蟹猴体内半衰期延长3.5倍。
4.3 糖基化修饰和电荷异质性对清除速率的影响糖基化是蛋白翻译后修饰的重要形式,目前大部分已批准上市的抗体药物都是糖基化修饰抗体[22]。研究表明,具有糖基化修饰的抗体分子表现出更优异的热稳定性。糖型是指抗体糖链上各个单糖之间的连接方式,常见糖型有双触角糖型(Di‑antennary)、单触角糖型(Monoantennary)、高甘露糖型(High mannose)、杂合糖型(Hybrid type)等(图1),携带不同糖型的抗体药物其体内清除速率有显著差异[23]。Falck 等[24]通过将4 种具有不同糖型的、经过糖基工程化的mAb 注射入大鼠体内,采用基于高效液相色谱串联质谱的糖蛋白组学方法研究具有不同糖型抗体的代谢规律,结果发现带有高甘露糖型和杂合糖型的抗体体内清除率增加。其中高甘露糖型抗体比带有常规双触角糖型的抗体清除率高出1.8~2.6倍;带有杂合糖型的抗体清除率比常规抗体高4.7 倍。此外,带有半乳糖基的单触角糖型抗体清除率也比常规抗体略有增加(1.2~1.4倍)。
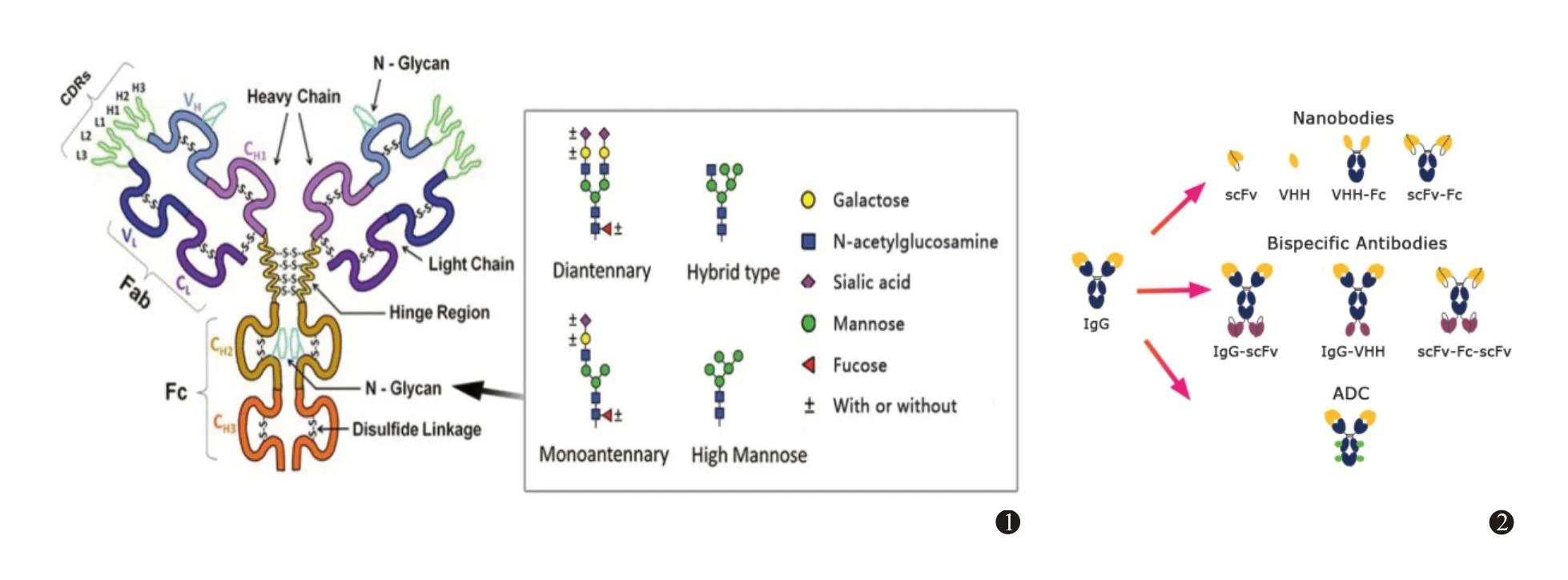
图1 经典的IgG型抗体结构 图2 由经典IgG结构抗体衍生得到纳米抗体、双特异性抗体、抗体药物偶联物的结构示意图
除了糖基化修饰以外,抗体药物还会发生诸如氧化、脱酰胺、氨基酸残基异构化等修饰,造成其氨基酸序列异质性,导致整体蛋白水平上的电荷异质性。抗体药物的电荷异质性直观体现为抗体蛋白分子等电点(pI)的差异,因此讨论电荷异质性对清除速率的影响等同于讨论抗体药物pI 对其体内半衰期的影响。通常情况下,氨基酸序列中含有较多碱性氨基酸的抗体具有较高的pI,而含有较多酸性氨基酸、发生脱酰胺作用或含有糖链中带有唾液酸的抗体具有较低的pI[25],各种在氨基酸水平的修饰都可能通过影响抗体的pI 进而影响抗体的体内清除。低pI 抗体由于其细胞摄取率低,体内半衰期更长;高pI 抗体表现出极高的黏附细胞表面阴离子位点的倾向,组织摄取增加,从循环中清除速率更快[26]。Igawa 等[27]在抗体药物Fab 区通过定点突变制备出不同pI 的抗体并对其PK 特征进行考察。实验结果显示,皮下和静脉内给药低pI 的突变体药物,清除率更低,清除率与pI 呈正相关。Ninad、Laird[28]在皮下注射抗体药物的PK 模型研究中发现,抗体的pI 可作为皮下注射后预测生物利用度和清除率的重要指标。此外,也有研究认为抗体药物pI 对于清除率的影响与抗体-FcRn 之间pH 依赖型的结合力强度有关,但具体影响的程度与机制还需要进一步研究与讨论[29]。
4.4 与抗药物抗体结合导致清除加快抗体药物作为异源蛋白质在一定程度上具有免疫原性,给药后会诱导机体产生抗药物抗体(ADA)。ADA的存在不仅会作为中和抗体导致抗体药物清除速率加快、药效降低,还会导致临床中出现药物过敏等严重不良反应[30]。人源化程度越高的抗体药物诱导产生ADA的倾向就越弱。然而,即便是人源化和完全人源化的抗体在应用过程中也无法完全避免ADA的产生[30]。
ADA 的产生可能引起抗体药物PK 特征发生改变,尤其是合并发生TMDD时,抗体药物的表观代谢趋势变得更加难以解释。由于TMDD的检测方式繁琐,而ADA的检测手段较为成熟便捷,因此在临床试验中若遇到PK异常的情况,最快速的排查方案就是首先测定血液中的ADA水平,再依次考虑其他潜在原因[16]。值得注意的是,许多已获批上市的抗体药物仍有着较高的诱导产生ADA的比例,并且不同种类抗体药物的ADA 发生比例也存在较大差异,约在0~75%之间[31]。Menter 等[32]在对于阿达木单抗(Adalimum‑ab)生物仿制药BI695501 的Ⅲ期临床数据进行报道时,特别指出原研药和生物仿制药给药后均在病人体内检出了ADA,检出阳性率均高达75%以上。
4.5 抗体药物的排泄完整的抗体药物分子由于具有较大的分子量,无法通过肾小球过滤排泄。抗体蛋白往往经由上述特定代谢途径,最终在细胞溶酶体内被多种酶剪切为小分子的氨基酸或短肽被机体吸收,重新进入机体氨基酸循环或经由肾脏排泄[16]。对于ADC,其释放出的小分子细胞毒性药物一般遵从小分子化合物的代谢及排泄规律,代谢产物经由尿液或粪便排出[33]。
4.6 新型抗体药物的药物代谢动力学相关研究进展随着生物工程技术的不断进步,更多新型抗体分子被研发出来,它们展现出比mAb更优的治疗效果,而全新的抗体结构也对药理学和PK研究带来了新的挑战。目前,所有获批的新一代抗体药物均采用静脉注射的给药途径,不涉及吸收问题,对于此类新型药物的PK研究多集中于靶向分布行为和代谢方式[34]。
Nb 的吸收及分布规律与经典mAb 一致,但Nb具有远小于经典抗体的分子量,导致其在体内被快速清除。尤其是分子量小于50 kDa 的Nb 可以经由肾小球的过滤作用被直接清除。因此,大多数治疗用Nb 药物在分子设计时会将ScFv 或VHH 结构与如Fc 片段等结构进行融合,通过FcRn 介导的再循环作用延长治疗性Nb 的体内半衰期[7,35]。Roy 等[36]构建了抗IL-6 的Nb(ALX-0061),该分子结构中具有Fc 片段,在食蟹猴体内的半衰期长达5.0~6.6 d,显著长于不具有Fc片段的结构。
BsAb 的PK 研究热点集中于探索其脑组织分布靶向性和代谢途径。传统的mAb 难以透过血脑屏障(BBB),无法实现对脑部疾病的治疗。BsAb 利用其多靶点的特性成功实现了抗体药物的脑内靶向。BsAb 的一个抗原结合臂可以与BBB 上特定受体相结合,使整个BsAb分子经由受体介导的胞吞作用跨越BBB,另一个抗原结合臂再引导进入脑组织内的BsAb 靶向至特定的病变部位发挥治疗作用[37]。Gadkar 等[38]建立了靶向TfR/BACE1 BsAb 的PK/药效动力学(PD)模型用于研究脑内靶向分布情况与BsAb抗原亲和力之间的关系,探讨了影响该抗体药物安全性与有效性的关键因素。
ADC可实现抗体药物和小分子化疗药物的优势互补,具有高度靶点选择性及扩大的治疗窗。ADC不仅比单独使用小分子化疗药物更长效,还具有靶向、低毒的特点。ADC偶联了小分子药物,常表现出相对于mAb更高的免疫原性,其诱导产生ADA加快体内清除的概率更高,PK 特征复杂[39]。ADC 与靶细胞表面抗原结合后被内化入细胞,在靶细胞内降解释放细胞毒性小分子药物从而杀伤靶细胞,这一过程中ADC的抗体部分在溶酶体内被降解,释放出的小分子化合物经由药物代谢酶介导的途径被降解排出[40]。ADC药物中小分子化疗药物在非靶组织释放所导致的毒性副作用一直是制约这一技术发展的瓶颈。因此,根据ADC的PK特点,研发设计安全长效的药物是未来的发展方向。Nagai等[41]优化了ADC中抗体与小分子化疗药物间的连接方式,将肽链作为化学接头,制备了连接有拓扑异构酶I抑制剂(DXd)的靶向HER2的ADC药物DS-8201a。临床前PK研究中发现,静脉给药后循环系统中DS-8201a 以完整分子的形式存在,DXd在靶组织内被特异释放且清除率高,成功实现了降低非靶组织内DXd暴露量的目的,使DS-8201a药物具有更好的安全性和有效性。
5 小结
抗体药物作为异质性较高的生物大分子,具有与小分子化学药物截然不同的PK 特点,尤其体现在代谢方式上,包括TMDD 效应、FcRn 介导的循环再利用、糖基化修饰和电荷异质性对清除速率的影响以及与ADA 结合导致清除加快等。在以上多重因素的共同作用下,抗体药物的体内代谢过程十分复杂。同时,随着制药领域推陈出新,如Nb、BsAb、ADC 等新型抗体药物开始逐渐进入研发与临床,对多种不同结构抗体药物PK 行为的研究就显得尤其重要。虽然现有的研究取得了一定成果,但迄今为止仍停留于对特定抗体药物PK 数据的报道,却缺少对现象形成机制的深入探讨。未来势必需要系统研究各种抗体药物PK 行为的内在机制,从而进一步理解不同结构抗体药物的体内作用机理,指导开发更加安全、有效的新一代抗体药物。
