芸薹根肿菌活细胞PMAxx-qPCR快速定量检测方法的建立与应用
2022-06-28李晓菁张思雨刘迪袁晓伟李兴盛石延霞谢学文李磊范腾飞李宝聚柴阿丽
李晓菁,张思雨,刘迪,袁晓伟,李兴盛,石延霞,谢学文,李磊,范腾飞,李宝聚,柴阿丽
芸薹根肿菌活细胞PMAxx-qPCR快速定量检测方法的建立与应用

1中国农业科学院蔬菜花卉研究所,北京 100081;2山东省华盛农业集团股份有限公司,山东青州 262500
【目的】由芸薹根肿菌()侵染引起的十字花科根肿病是一种世界性土传病害,病原菌长期存在于土壤中,对十字花科作物造成严重威胁。改良叠氮溴化丙锭(propidium monoazide xx,PMAxx)可选择性地穿透受损的死细胞膜,并抑制死细胞DNA的实时荧光定量PCR(qPCR)扩增。本文将PMAxx与qPCR技术相结合,建立一种快速检测芸薹根肿菌活菌的方法,为根肿病的早期诊断及制定科学的防控措施提供依据。【方法】配置浓度分别为0、5、10、20、40、60 µmol·L-1的叠氮溴化丙锭PMA和改良叠氮溴化丙锭PMAxx,比较两种核酸染料对芸薹根肿菌死细胞DNA扩增的抑制效果,确定最佳核酸染料及工作浓度;设置光照时间分别为0、2、5、10、15和20 min,进行最佳光照时间的优化,建立芸薹根肿菌活细胞PMAxx-qPCR快速检测体系。设置芸薹根肿菌活孢子百分比为0、0.01%、0.1%、1%、10%、25%、50%、75%和100%的混合体系,验证PMAxx-qPCR体系的准确性,并应用于田间土壤样本中芸薹根肿菌活孢子的定量检测。【结果】PMAxx对芸薹根肿菌死细胞DNA的扩增抑制效果更好,当芸薹根肿菌浓度为1×108个孢子/mL,PMAxx预处理的最适终浓度为4 µmol·L-1,最佳光照时间为10 min时,可有效地抑制死孢子DNA的扩增,仅以有活力孢子DNA为靶标选择性地扩增。利用PMAxx-qPCR技术检测已知不同活孢子比例的菌悬液样品,各样品实测孢子存活率和理论存活率之间呈正相关(2=0.992)。对田间采集的25份土壤样本,采用PMAxx-qPCR方法检测到11份样本中携带芸薹根肿菌,活细胞DNA浓度为32.35—6.97×103fg·g-1。【结论】建立了基于PMAxx-qPCR的芸薹根肿菌活细胞定量检测技术,该技术具有快速、准确、灵敏的特点,解决了qPCR不能仅对活体病原菌进行准确鉴别和定量分析的问题,为制定有效的根肿病防控策略提供了依据。
芸薹根肿菌;根肿病;改良叠氮溴化丙锭;实时荧光定量PCR;活性孢子
0 引言
【研究意义】十字花科根肿病是由芸薹根肿菌()侵染引起的一种世界范围内的土传病害[1-2],大白菜、甘蓝、青花菜、花椰菜、油菜等十字花科作物均可受害,引起产量下降,甚至造成绝产绝收,严重影响十字花科产业发展[3-5]。近年来,随着十字花科作物在我国广泛种植,根肿病的影响越来越大,并缺乏有效的防治手段[6]。病原菌可以在土壤中长期存活,最长可达20年,一旦土壤中带有根肿菌,将不适宜种植十字花科作物[7-8]。因此,建立高效、灵敏的芸薹根肿菌活细胞定量检测方法,对于病害的早期诊断以及制定科学有效的防控措施防止该病菌传播十分重要。【前人研究进展】目前,针对芸薹根肿菌的检测大多采用分子生物学技术,如普通PCR[9-12]、巢式PCR[13-14]、实时荧光定量PCR(real-time fluorescent quantitative PCR,qPCR)[15-20]等。然而,普通PCR和巢氏PCR都只能进行定性检测[9-14],不能进行定量检测。qPCR技术的发展为从基因水平上定量监测各种微生物提供了十分有效的手段。Wallenhammar等分别利用特异性引物建立了土壤中芸薹根肿菌的qPCR检测体系,检出下限为103—104个孢子/g土壤[16-17]。然而,qPCR方法检测的是土壤中病原菌的总量,不仅能扩增有活性芸薹根肿菌孢子DNA,也会扩增死孢子DNA以及游离状态的病原菌DNA,因此不能区分样品中芸薹根肿菌的存活状态,造成芸薹根肿菌活菌含量的高估[21]。叠氮溴化丙锭(propidium monoazide,PMA)是一种特异的活性染料,可以穿透死细胞不完整的细胞膜,与其DNA进行不可逆的共价结合,被这种活性染料结合的DNA不能进行PCR扩增,从而阻止死细胞DNA的扩增,达到检测活性细胞的目的[22]。PMA-qPCR已经应用于医学、食品等方面病原真菌、细菌和病毒活性的定量检测[23-27],在植物病原菌检测方面,该技术已经用于西瓜细菌性果斑病菌()、黄瓜细菌性角斑病菌(pv.)、番茄细菌性斑点病菌(pv.)、番茄溃疡病菌(subsp.)等病原细菌活细胞的定量检测[28-32]。PMAxx是PMA的改良结构,同样可以穿透死菌或者受损菌的细胞膜并与其DNA进行结合,在强光条件下使DNA构象发生改变,从而抑制PCR扩增;与PMA相比,PMAxx能更高效抑制死菌DNA的扩增,从而具有更高效区分有活性与无活性病原菌的能力[31]。【本研究切入点】为解决现有qPCR技术无法判断病原菌活性,不能实现土壤中有活性病原菌准确检测和病害预警的问题,本文将PMAxx与qPCR技术相结合,建立一种快速、准确的定量检测芸薹根肿菌活细胞的方法。【拟解决的关键问题】优化PMAxx预处理最佳浓度和光照时间,建立基于PMAxx-qPCR的芸薹根肿菌活细胞定量检测体系,解决qPCR不能区分病原菌活性的问题。
1 材料与方法
试验于2019年1月至2021年5月在中国农业科学院蔬菜花卉研究所完成。
1.1 供试菌株和种子
供试芸薹根肿菌采自云南昆明的大白菜发病肿根,保存于中国农业科学院蔬菜花卉研究所蔬菜病害综合防治课题组,保存条件为-20℃冰箱。大白菜‘中白60’由中蔬种业科技(北京)有限公司提供。
1.2 主要试剂和设备
PMA和PMAxx(美国,Biotum公司)购自北京博晟思源生物科技有限公司;ABI 7500实时荧光定量PCR仪购自美国ABI公司;Bio-Rad S1000 PCR仪购自美国伯乐公司。
1.3 基因组DNA提取和引物特异性检测
采用改良CTAB法,提取肿根组织中芸薹根肿菌DNA和11种非靶标病原菌(表1)基因组DNA。使用课题组前期筛选的芸薹根肿菌特异性引物PBF3(5′-TCTTGCGTGTCGCTGTATTC-3′)和PBR3(5′-ATAGGTTGGGGTAACTTGGC-3′)[18],对靶标和非靶标菌的基因组DNA进行qPCR扩增,以健康白菜根组织DNA和ddH2O为阴性对照。qPCR反应体系(20 μL):SYBR qPCR Master Mix(博迈德,北京)10 μL,上、下游引物各0.4 μL,DNA模板2 μL。qPCR扩增程序:95℃ 10 min;95℃ 15 s,60℃ 34 s,共40个循环。反应结束后分析扩增曲线及熔解曲线,引物序列由北京博迈德科技有限公司合成。

表1 引物特异性检测所用菌株
1.4 质粒标准品制备
将引物PBF3/PBR3普通PCR扩增到的片段,采用离心柱型琼脂糖凝胶回收试剂盒(全式金,北京)进行回收纯化。PCR反应体系(50 μL):上、下游引物各1 μL,2×Taq Master Mix 25 μL,模板DNA 2 μL。PCR扩增程序:94℃3 min;94℃1 min,60℃1 min,72℃1 min,共38个循环;72℃10 min。将目标DNA与pEASY®-T1克隆载体(全式金,北京)连接,转化大肠杆菌Trans1-T1感受态细胞(全式金,北京)。取200 μL菌液涂布在含有100 μg·mL-1氨苄青霉素的LB固体培养基上,置于37℃恒温培养室倒置培养24 h。随机挑取一定数量的白色单菌落,在含100 μg·mL-1氨苄青霉素的LB液体培养基中振荡培养。PCR鉴定为阳性克隆后,随机选取10个阳性克隆,送北京博迈德生物公司测序。
1.5 芸薹根肿菌qPCR检测体系建立
使用Thermo Scientific NanoDrop 2000c紫外分光光度计,测定芸薹根肿菌质粒DNA浓度,将初始浓度16.12 ng·μL-1做10倍梯度稀释16.12—1.612×10-6ng·μL-1,建立qPCR标准曲线。以qPCR扩增时所得循环数Ct值为纵坐标轴,以起始质粒DNA浓度的对数为横坐标轴,绘制芸薹根肿菌qPCR标准曲线,并计算回归方程。
1.6 芸薹根肿菌活孢子和死孢子悬浮液制备
取新鲜发病肿根,加入适量无菌水,用组织捣碎机打碎至匀浆状,8层纱布过滤。用血球计数板调节孢子悬浮液浓度,制备成1×108个休眠孢子/mL的活孢子菌悬液备用。取活孢子悬浮液,100℃沸水浴中处理30 min高温致死,振荡5 min使致死成团的休眠孢子均匀分散,制备成1×108个休眠孢子/mL的死孢子菌悬液备用。
1.7 PMA和PMAxx的效果比较
PMA和PMAxx母液制备和储存:将1 mg PMA溶于98 μL的20%二甲基亚砜(DMSO)溶液,制成浓度为20 mmol·L-1的母液,-20℃黑暗储存。订购20 mmol·L-1的PMAxx,-20℃黑暗储存。
PMA和PMAxx的抑制效果比较:取2 mL离心管,将不同体积PMA和PMAxx分别加入1 mL浓度为108个孢子/mL的活孢子和死孢子悬浮液中,使PMA和PMAxx终浓度分别为0、5、10、20、40和60 μmol·L-1,充分混匀。锡纸包裹离心管,置于28℃、180 r/min避光振荡处理30 min,使PMA和PMAxx与死孢子充分接触。之后拿掉锡纸,置于冰上,在距离50 W卤钨灯20 cm处光照冰浴20 min,激活PMA和PMAxx,使其与死菌DNA充分结合。处理后的菌悬液,离心弃上清,使用法医样品DNA提取试剂盒(Omega公司)提取芸薹根肿菌沉淀DNA,进行qPCR扩增。以未经PMA或PMAxx处理的菌悬液DNA为阳性对照,ddH2O为阴性对照。每个处理设置4个平行样,每个DNA模板qPCR反应进行3次重复。通过计算dCt值和ddCt值,筛选光敏染料。dCt值为光敏染料处理样品的Ct值减去未经染料处理样品的Ct值,而ddCt值为死孢子的dCt值减去活孢子的dCt值[31]。
1.8 最适PMAxx终浓度筛选
将PMAxx加入含有1 mL浓度108个孢子/mL的活孢子悬浮液和高温致死菌悬液的2 mL离心管中,使PMAxx终浓度分别达到0、0.2、0.5、1、1.5、2、4、6、8和10 μmol·L-1,避光振荡处理30 min后,光照冰浴20 min。之后按照1.7方法提取DNA,进行qPCR扩增。每个处理设置4个平行样,每个qPCR反应进行3次重复。
1.9 最适PMAxx光照时间筛选
将PMAxx加入含有1 mL浓度108个孢子/mL的活孢子悬浮液和高温致死菌悬液的2 mL离心管中,使PMAxx终浓度为4 μmol·L-1。避光振荡处理30 min后,在距离50 W卤钨灯20 cm处光照冰浴孵育,光照时间分别为0、2、5、10、15和20 min,进行PMAxx最佳光照时间的优化。之后按照1.7方法提取DNA,进行qPCR扩增。每个处理设置4个平行样,每个qPCR反应进行3次重复。
1.10 PMAxx-qPCR检测体系准确性验证
取浓度108个孢子/mL芸薹根肿菌活孢子悬浮液和死孢子悬浮液,按不同体积比混合,分别配制成活孢子比例为0、0.01%、0.1%、1%、10%、25%、50%、75%和100%的死/活菌混合体系。每个处理总体积10 mL,分别进行qPCR和PMAxx-qPCR检测。每个比例设置4个平行样,每个qPCR反应进行3次重复。qPCR检测:不使用PMAxx预处理,按照1.7方法提取DNA,进行qPCR扩增。PMAxx-qPCR检测:首先对不同活孢子比例的菌悬浮液进行PMAxx预处理,PMAxx处理浓度为4 μmol·L-1,光照时间为10 min,然后进行qPCR扩增。根据检测获得的Ct值,计算菌悬液浓度,分析比较实际活孢子率和理论活孢子率的差异及其相关性,验证PMAxx-qPCR检测体系的准确性。
1.11 PMAxx-qPCR检测体系应用
2019—2020年,从湖北恩施、山东青岛、江苏无锡、辽宁沈阳、河南南阳、四川绵阳和广元等十字花科蔬菜主产区采集田间土壤样本25份。每份样品各取5 g,加入20 mL无菌水,旋涡振荡器振荡混匀,8层纱布过滤去除土壤颗粒及杂质,获得的土壤菌悬液分别进行qPCR和PMAxx-qPCR检测,分析土样中根肿菌总量和有活力根肿菌量。同时,将采集的土样装入6 cm×6 cm育苗钵中,种植大白菜,45 d后调查根肿病发病情况。根肿病调查及分级标准:0级为根部无肿大;1级肿根只附着在侧根上,占全部根系1%—25%;2级主根上有肿根附着,侧根上肿根占26%—50%;3级主根上有肿根附着,肿根占根系51%—75%;4级主根上有根肿附着,肿根占根系75%以上[16]。病情指数=∑(各级病株数×该级代表值)/(调查总株数×4)×100。
2 结果
2.1 引物特异性分析
以健康白菜根组织DNA和ddH2O为阴性对照,利用引物对PBF3/PBR3对靶标芸薹根肿菌和11种非靶标病原菌基因组DNA进行qPCR扩增。在qPCR检测中,只有Ct介于11—29,同时具有单一熔解曲线峰值,才被认为是有效结果。芸薹根肿菌Ct平均值为12.32(图1),熔解曲线获得单一峰值,Tm值为84.75℃(图2);而非靶标菌株、健康白菜根组织DNA和ddH2O的Ct值均大于32,同时也没有获得熔解曲线或出现不同的Tm值。说明引物对PBF3/PBR3在qPCR检测中具有较高的特异性。

1:芸薹根肿菌P. brassicae;2:尖镰孢F. oxysporum;3:茄镰孢F. solani;4:半裸镰孢F. incarnatum;5:立枯丝核菌R. solani;6:核盘菌S. sclerotiorum;7:芸薹链格孢A. brassicae;8:长孢轮枝菌V. longisporum;9:瓜果腐霉P. aphanidermatum;10:灰葡萄孢B. cinerea;11:胡萝卜软腐果胶杆菌胡萝卜亚种P. carotovorun subsp. carotovorum;12:野油菜黄单胞菌野油菜致病变种X. campestris pv. campestris;13:健康大白菜根组织Root tissue of healthy Chinese cabbage;14:ddH2O
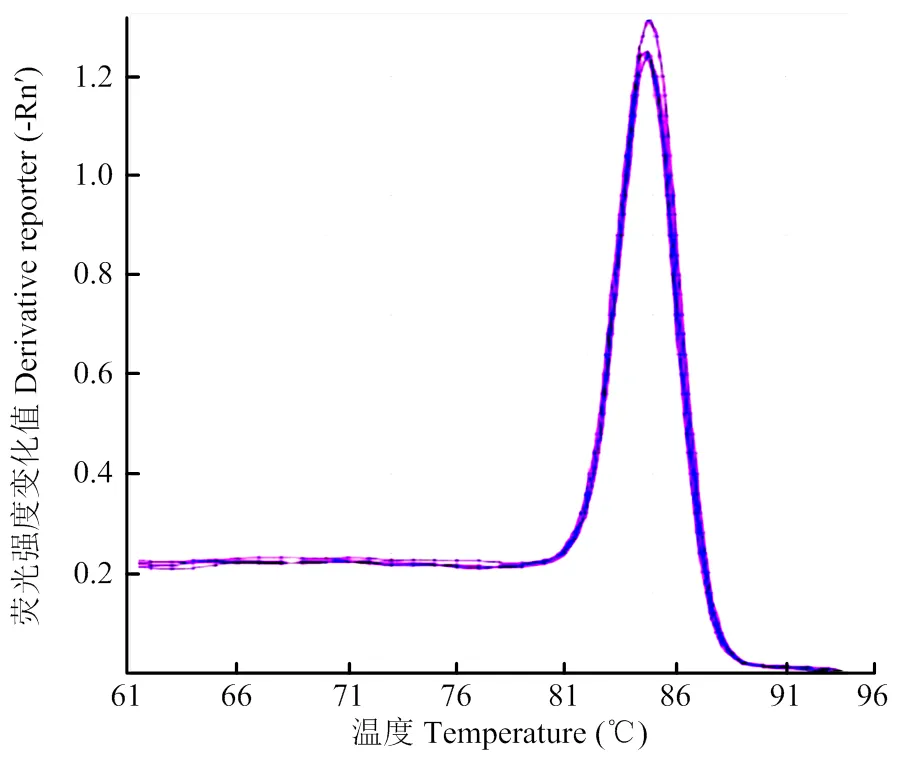
具有单一熔解曲线峰值的菌株为芸薹根肿菌,单一Tm值84.75℃;而非靶标菌株、健康白菜根组织DNA和ddH2O均没有明显峰值出现The melting curve of P. brassicae had single peak with Tm 84.75℃. The melting curve of non-target strains, root tissue of healthy Chinese cabbage and ddH2O had no peak or slightly peaks with different Tm values
2.2 芸薹根肿菌qPCR检测体系标准曲线建立
将芸薹根肿菌质粒DNA进行10倍梯度稀释16.12—1.612×10-6ng·μL-1,以不同稀释倍数的质粒DNA作为模板,构建qPCR标准曲线。以质粒DNA浓度对数值为X轴,循环数Ct值为Y轴,绘制标准曲线,并计算回归方程。扩增反应循环数Ct值与质粒DNA浓度的对数值之间有良好的线性关系:=-3.6012+34.314,相关系数²=0.9974(图3)。
2.3 PMA和PMAxx检测效果比较
当PMA和PMAxx浓度为0 μmol·L-1时,芸薹根肿菌活孢子和死孢子悬浮液qPCR检测Ct值分别为14.53和14.86,差异不显著。随着PMA和PMAxx浓度增加,活孢子悬浮液Ct值轻微增大,而死孢子悬浮液Ct值显著增大,说明PMA及PMAxx均对死孢子DNA产生抑制扩增的效果。当PMA和PMAxx浓度达到60 μmol·L-1时,活孢子Ct值分别为18.52和18.58,差异不显著;而PMAxx处理死孢子的Ct值32.61显著高于PMA处理的Ct值29.84。在各浓度下,PMAxx处理的ddCt值均高于PMA处理(表2),说明PMAxx对死孢子基因组DNA扩增的抑制效果强于PMA。因此,选择PMAxx作为核酸染料,构建PMAxx-qPCR芸薹根肿菌活孢子定量检测体系。
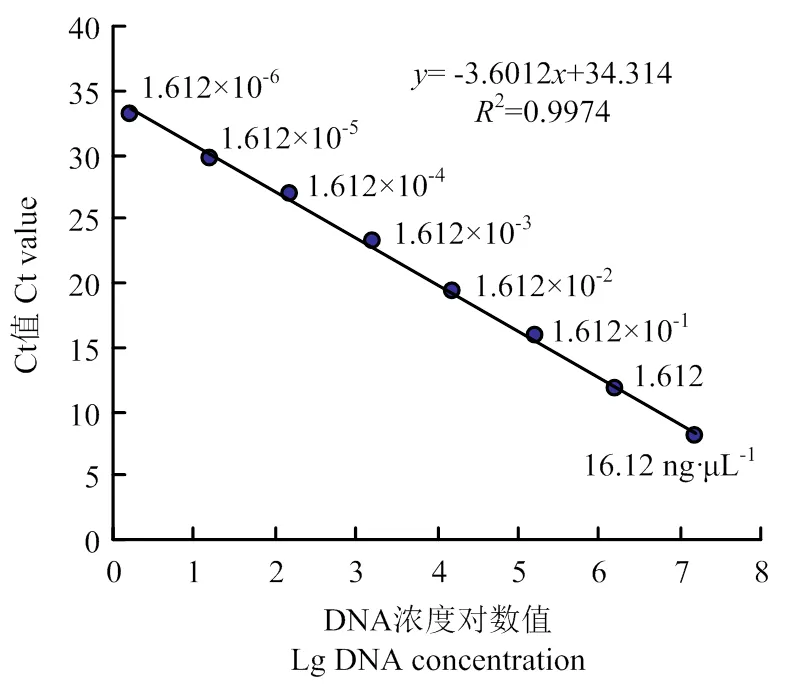
图3 芸薹根肿菌荧光定量PCR标准曲线
2.4 最适PMAxx终浓度
采用终浓度0、0.2、0.5、1、1.5、2、4、6、8和10 μmol·L-1的PMAxx,对108个孢子/mL的芸薹根肿菌活孢子和死孢子悬浮液进行处理。当PMAxx浓度低于4 μmol·L-1时,死孢子检测的Ct值随PMAxx处理浓度的升高而逐渐增大。而当PMAxx浓度大于4 μmol·L-1时,随着PMAxx浓度增大,死孢子DNA的扩增受到了显著抑制,qPCR检测的Ct值无显著变化,说明PMAxx浓度为4 μmol·L-1可最大限度地抑制死孢子DNA扩增。同时,不同浓度PMAxx处理活孢子悬浮液,当PMAxx浓度低于4 μmol·L-1时,PMAxx浓度对其qPCR扩增无显著抑制作用;但当PMAxx浓度高于4 μmol·L-1,其Ct值略有增加,说明高浓度PMAxx影响了活孢子的检测。因此,PMAxx的最适终浓度为4 μmol·L-1,该浓度的PMAxx既可以显著抑制死孢子的扩增,又对活孢子扩增没有显著影响(图4-A)。
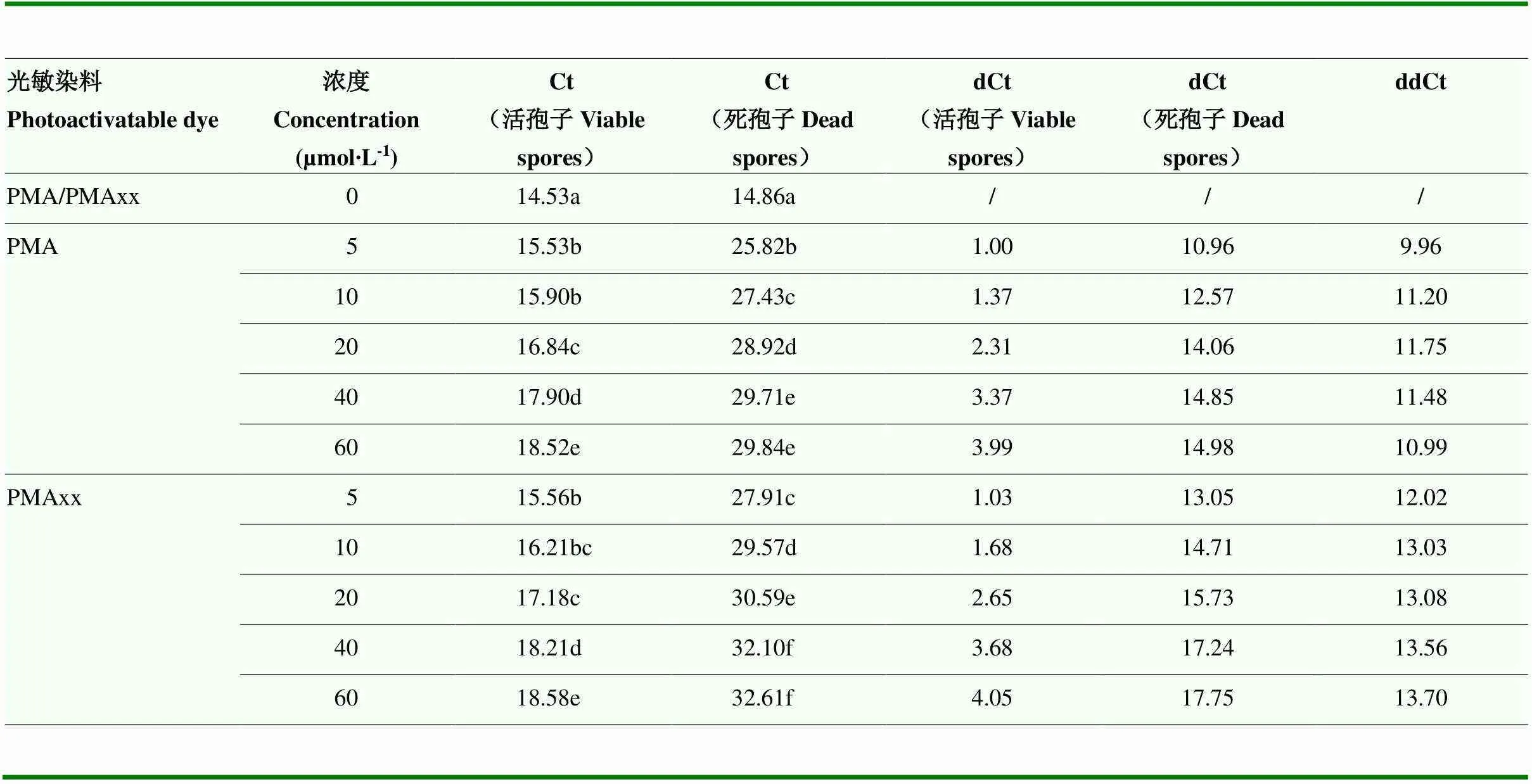
表2 PMA和PMAxx预处理芸薹根肿菌qPCR检测的Ct值
ddCt = dCt(死孢子Dead spores)-dCt(活孢子Viable spores);dCt(死孢子Dead spores)= Ct(光敏染料处理死孢子Dead spores with dye)-Ct(无染料处理死孢子Dead spores without dye);dCt(活孢子Viable spores)= Ct(光敏染料处理活孢子Viable spores with dye)-Ct(无染料处理活孢子Viable spores without dye)
2.5 最适PMAxx光照时间
使用PMAxx预处理芸薹根肿菌活孢子和死孢子悬浮液,当PMAxx预处理0 min时,PMAxx没有发生光化反应,无法抑制死孢子DNA的扩增,活孢子qPCR反应的Ct值与死孢子Ct值差异不显著。当PMAxx预处理2、5和10 min时,PMAxx产生光化学反应,抑制死孢子DNA扩增,随着光处理时间延长,死孢子qPCR反应的Ct值增加,而活孢子Ct值无显著差异;当光处理10、15和20 min时,死孢子Ct值qPCR反应差异不显著。因此,PMAxx最适光照时间为10 min,PMAxx能够完全与死孢子DNA共价联结(图4-B)。
2.6 芸薹根肿菌PMAxx-qPCR检测体系准确性验证
为了验证PMAxx-qPCR体系的可靠性,对不同比例浓度为108个孢子/mL的芸薹根肿菌活孢子和死孢子混合悬浮液(0.01%、0.1%、1%、10%、25%、50%、75%和100%活孢子)分别采用qPCR和PMAxx-qPCR进行检测。
使用qPCR检测,无论活孢子比例是否变化,Ct值始终维持在稳定水平,无显著差异(>0.05)。当活孢子比例从100%降至0.01%时,qPCR的Ct值介于10.04—10.81,对应DNA浓度对数值在6.57—6.74。对于PMAxx-qPCR法,随着活孢子比例下降,Ct值也逐渐増加。当活孢子比例由100%逐渐降至0.01%时,Ct值由10.34升至23.59,对应DNA浓度对数值由6.66降至2.98(表3)。PMAxx-qPCR检测的芸薹根肿菌基因组DNA浓度的对数值与理论活孢子浓度的对数值之间存在良好的线性关系:=0.8981-0.7148,2=0.992(图5)。结果表明,PMAxx-qPCR可以有效区分菌悬液中的活孢子和死孢子,选择性地对活孢子DNA进行qPCR扩増,而qPCR法无法区分活孢子和死孢子,其检测结果会对活孢子浓度造成高估。
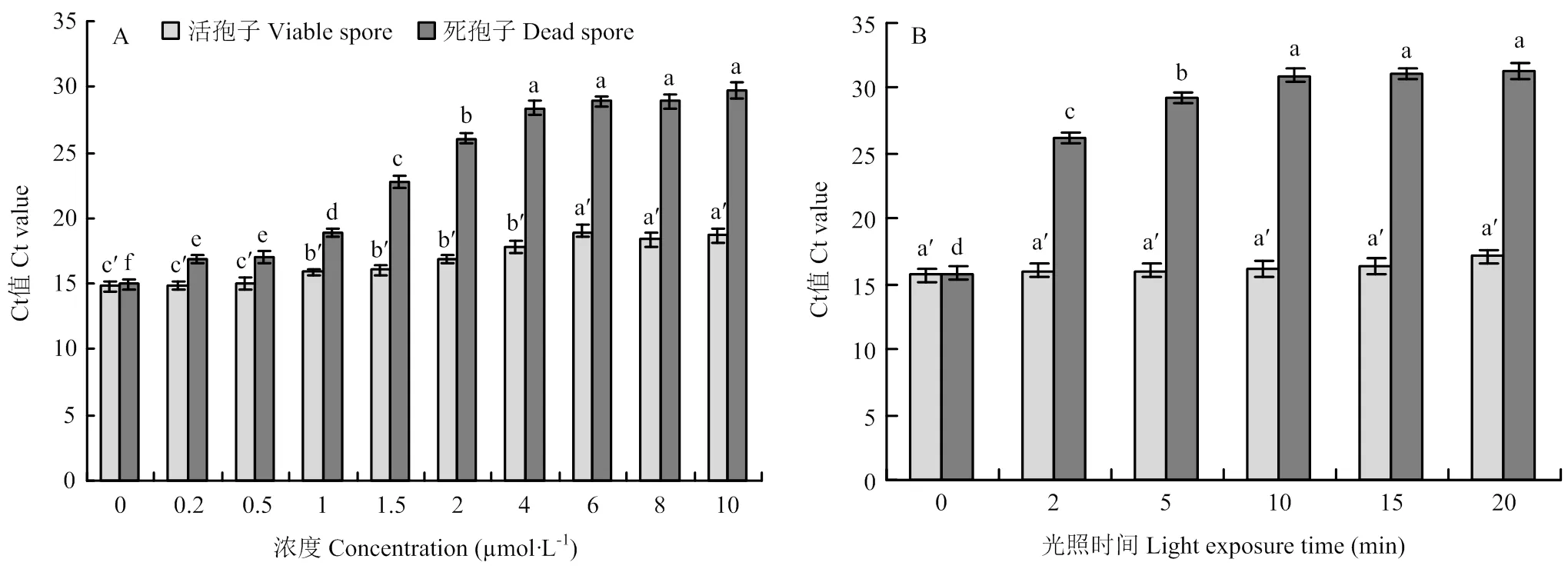
图4 PMAxx浓度(A)和光照时间(B)对芸薹根肿菌活孢子和死孢子DNA扩增的影响

表3 不同比例芸薹根肿菌活孢子悬浮液qPCR和PMAxx-qPCR检测结果
数据为平均值±标准差,不同小写字母表示在0.05水平上差异显著
Data were presented as mean±SD, data with different lowercase letters indicated significant differences at 0.05 level
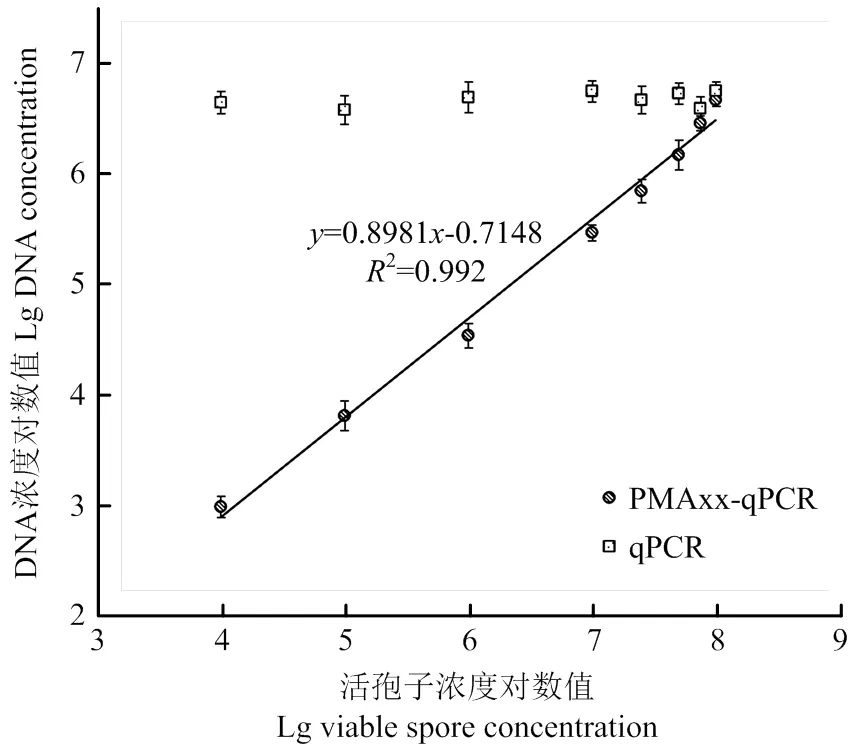
图5 不同比例芸薹根肿菌活孢子悬浮液PMAxx-qPCR检测准确性验证
2.7 PMAxx-qPCR检测体系应用
使用qPCR和PMAxx-qPCR方法对采集的25份田间肿根土壤样本进行检测,其中11份样本检测到芸薹根肿菌。qPCR检测Ct值介于17.00—25.21,对应土壤中芸薹根肿菌DNA浓度为3.37×102—6.44×104fg·g-1;而PMAxx-qPCR检测Ct值高于qPCR检测的Ct值,介于20.47—28.88,对应有活性芸薹根肿菌DNA浓度为32.35—6.97×103fg·g-1。除采自山东青岛的土壤外,其余10份田间土壤样本种植大白菜后,均发生根肿病。不同土壤样本中检测到的芸薹根肿菌活孢子量不同,其中,采自湖北恩施的土壤样本中含有的活孢子带菌量最多,为6.97×103fg·g-1,种植大白菜后,根肿病的病情指数最高为50.48;采自山东青岛的土壤样本中含有的活孢子带菌量最少,为32.35 fg·g-1,种植大白菜后未发生根肿病(表4)。
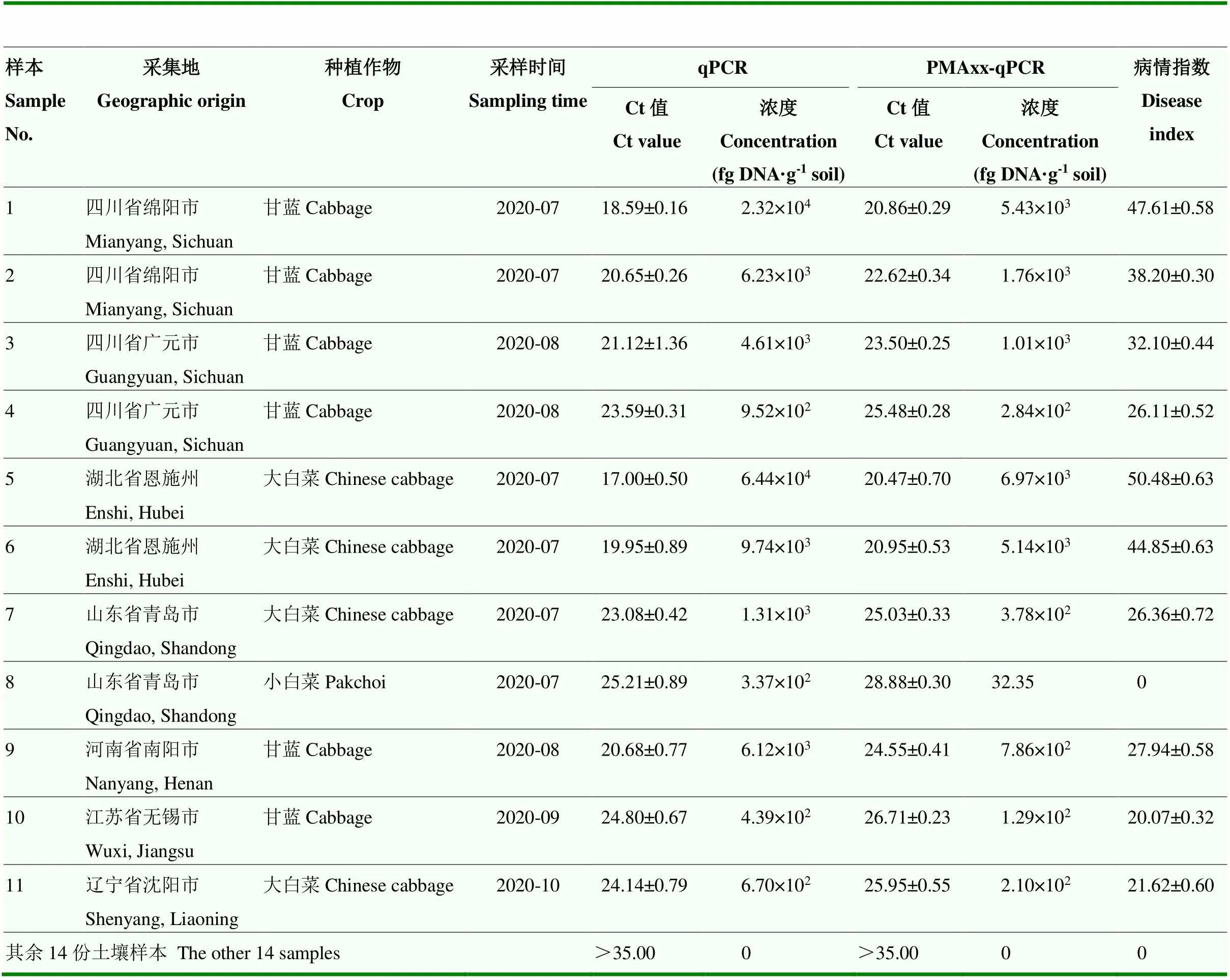
表4 十字花科根肿病田间发病土壤带菌量qPCR及PMAxx-qPCR检测结果
3 讨论
3.1 土壤中芸薹根肿菌活细胞PMAxx-qPCR定量检测方法
我国十字花科蔬菜种类多、分布广、种植面积大,根肿病常年危害面积高达320—400万公顷,占十字花科作物种植面积的1/3以上,严重制约着十字花科产业的发展[6,10,15]。根肿病的主要侵染来源[7-8],为有效预防根肿病的发生蔓延,对病原菌的早期、快速检测尤为重要。
本研究将PMAxx预处理与qPCR相结合,建立了PMAxx-qPCR检测芸薹根肿菌活细胞的方法,并用于田间土壤中根肿菌活细胞的定量检测,解决了qPCR技术不能判断土壤中病原菌活性,容易造成目标病菌初侵染量高估的问题[21],检测时间由传统生物测定方法的30—45 d[17]缩短至1—3 h,解决了传统方法检测周期长、准确性差、检出率低等难题,为根肿病的早期预防和精准治疗提供了技术支持。
3.2 PMAxx预处理条件优化
对于不同的病原菌及浓度,区分细胞活性的最适PMAxx预处理条件不同。本研究对芸薹根肿菌PMAxx预处理的最适终浓度和光照时间进行了优化。PMAxx预处理浓度是影响区分活孢子和死孢子水平的重要因子。理想的PMAxx浓度应该既保证与死孢子DNA充分共价结合,又不影响活孢子DNA的PCR扩增。使用不同浓度的PMAxx对1×108个孢子/mL芸薹根肿菌菌悬液进行处理,并对处理后的菌悬液进行qPCR检测。当PMAxx浓度低于4 μmol·L-1时,低浓度的PMAxx不能完全与死菌体DNA结合,无法充分抑制死孢子DNA的扩增,检测的Ct值随PMAxx浓度升高而逐渐增大,而当PMAxx浓度大于4 μmol·L-1时,死孢子DNA的扩增受到了显著抑制,因此,抑制死细胞扩增的PMAxx最适终浓度为4 µmol·L-1。
PMAxx处理分为暗反应和光反应两个阶段,在暗反应阶段,PMAxx渗透进入死孢子,与死孢子DNA充分接触;在光反应阶段,PMAxx与死孢子DNA发生不可逆的共价联结,后续PCR扩增过程中抑制死孢子DNA的扩增。暗反应阶段处理时间越长,PMAxx渗透越充分,本试验避光处理时间设置为30 min,以便PMAxx与死孢子DNA充分结合。光反应阶段,PMAxx光照预处理10 min后,PMAxx可以与DNA进行完全的共价结合,从而抑制死细胞DNA的扩增,而多余的PMAxx在光照处理10 min后,其光化学活性也会完全消失,不会影响qPCR过程,仅以有活力孢子DNA为靶标选择性地扩增活菌。
3.3 PMAxx-qPCR检测技术的应用
对已知活孢子比例的芸薹根肿菌混合体系进行检测,无论活孢子比例是否变化,qPCR检测的Ct值始终维持在稳定水平。然而,随着活孢子比例下降,PMAxx-qPCR测得的DNA浓度对数值也逐渐降低,并且与理论活孢子浓度对数值之间存在良好的线性关系。说明本研究建立的PMAxx-qPCR方法可以有效区分芸薹根肿菌活孢子和死孢子。对田间收集的土壤样本进行检测,在25份样本中有11份采自发病地块的土壤样本检测到携带芸薹根肿菌,根肿菌活细胞DNA浓度为32.35—6.97×103fg·g-1,而采自健康田块的14份土壤样本未检测到根肿菌。盆栽试验结果表明,当PMAxx-qPCR检测土壤中根肿菌活细胞DNA浓度大于1.29×102fg·g-1时,有导致根肿病暴发的潜在危险。因此,PMAxx-qPCR检测技术可以作为一种有效的工具用于十字花科蔬菜病害的早期诊断和预警。
4 结论
建立了基于PMAxx预处理结合qPCR的芸薹根肿菌活细胞定量检测技术,确定了PMAxx的最适终浓度和处理时间,该技术可以快速、准确、定量检测菌悬液和土壤中的芸薹根肿菌并区分病原菌的活性,准确反映实际样本中带活菌的情况,对于根肿病的预防和病原菌的早期诊断具有潜在的应用价值。
[1] DIXON G R. The occurrence and economic impact ofand clubroot disease. Journal of Plant Growth Regulation, 2009, 28(3): 194-202.
[2] Howard R J, Strelkov S E, Harding M W. Clubroot of cruciferous crops - new perspectives on an old disease. Canadian Journal of Plant Pathology, 2010, 32(1): 43-57.
[3] Devos S, Vissenberg K, Verbelen J P, Prinsen E. Infection of Chinese cabbage byleads to a stimulation of plant growth: impacts on cell wall metabolism and hormone balance. New Phytologist, 2005, 166(1): 241-250.
[4] Hwang S F, Strelkov S E, Feng J, Gossen B D, Howard R J.: a review of an emerging pathogen of the Canadian canola () crop. Molecular plant pathology, 2012, 13(2): 105-113.
[5] Tso H H, Galindo-Gonzalez L, Strelkov S E. Current and future pathotyping platforms forin Canada. Plants, 2021, 10(7): 1446.
[6] Chai A L, Xie X W, Shi Y X, Li B J. Research status of clubroot () on cruciferous crops in China. Canadian Journal of Plant Pathology, 2014, 36: 142-153.
[7] Donald C, Porter I. Integrated control of clubroot. Journal of Plant Growth Regulation, 2009, 28: 289-303.
[8] Tsushima S. Perspective of integrated pest management - A case study: Clubroot disease of crucifers. Journal of pesticide science, 2000, 25(3): 296-299.
[9] Ito S, Maehara T, Maruno E, Tanaka S, Kameya-Iwaki M, Kishi F. Development of a PCR-based assay for the detection ofin soil. Journal of Phytopathology, 1999, 147(2): 83-88.
[10] 杨佩文, 杨勤忠, 王群, 李家瑞, 曾莉. 十字花科蔬菜根肿病菌的PCR检测. 云南农业大学学报, 2002, 17(2): 137-139, 157.
Yang P W, Yang q z, wang q, Li J r, zeng L. PCR detection ofcausing cruciferae clubroot. Journal of Yunnan Agricultural University, 2002, 17(2): 137-139, 157. (in Chinese)
[11] Cao T, Tewari J, Strelkov S E. Molecular detection of, causal agent of clubroot of crucifers, in plant and soil. Plant Disease, 2007, 91(1): 80-87.
[12] Faggian R, Strelkov S E. Detection and measurement of. Journal of Plant Growth Regulation, 2009, 28: 282-288.
[13] FAGGIAN R, BULMAN S R, LAWRIE A C, PORTER I J. Specific polymerase chain reaction primers for the detection ofin soil and water. Phytopathology, 1999, 89(5): 392-397.
[14] Wallenhammar A C, Arwidsson O. Detection ofby PCR in naturally infested soils. European Journal of Plant Pathology, 2001, 107(3): 313-321.
[15] 李淼, 周丽洪, 刘雅婷, 刘峰, 杨俊, 姬广海. 云南省十字花科蔬菜根肿病的实时荧光定量PCR检测. 云南农业大学学报(自然科学), 2016, 31(1): 43-48.
Li m, Zhou l h, Liu Y T, Liu f, yang j, ji g h. Detection ofwith real-time quantitative PCR in Yunnan province. Journal of Yunnan Agricultural University (Natural Science), 2016, 31(1): 43-48. (in Chinese)
[16] Wallenhammar A C, Almquist C, Söderström M, Jonsson A. In-field distribution ofmeasured using quantitative real-time PCR. Plant Pathology, 2012, 61(1): 16-28.
[17] Li J P, Li Y, Shi Y X, Xie X w, Chai A l, Li B j. Development of a real-time PCR assay forand its detection in soil samples. Journal of Integrative Agriculture, 2013, 12(10): 1799-1806.
[18] Chai A L, Li J P, Xie X W, ShiYX,Li BJ. Dissemination ofin livestock manure detected by qPCR. Plant Pathology, 2016, 65(1): 137-144.
[19] Wen R, Lee J, Chu M, Tonu N, Dumonceaux T, Gossen B D, Yu F, Peng GQuantification ofresting spores in soils using droplet digital PCR (ddPCR). Plant Disease, 2020, 104(4): 1188-1194.
[20] Rennie D C, Manolii V P, Cao T, Hwang S F, Howard R J, Strelkov S E. Direct evidence of surface infestation of seeds and tubers byand quantification of spore loads. Plant Pathology, 2011, 60(5): 811-819.
[21] Al-Daoud F, Gossen B D, Robson J, McDonald M R. Propidium monoazide improves quantification of resting spores ofwith qPCR. Plant Disease, 2017, 101(3): 442-447.
[22] NockerA, CamperAK. Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Applied and Environmental Microbiology, 2006, 72(3):1997-2004.
[23] LiuY, MustaphaA. Detection of viableO157:H7 in ground beef by propidium monoazide real-time PCR.International Journal of Food Microbiology, 2014, 170: 48-54.
[24] WangS, LevinRE. Discrimination of viablecells from dead cells in real-time PCR. Journal of Microbiological Methods, 2006, 64(1):1-8.
[25] YÁÑEZMA, NOCKERA, SORIA-SORIAE, MÚRTULAR, MARTÍNEZL, CATALÁNV. Quantification of viablecells using propidium monoazide combined with quantitative PCR.Journal of Microbiological Methods, 2011, 85(2):124-130.
[26] CRESPO-SEMPERE A, ESTIARTE N, MARÍN S, SANCHIS V, RAMOS AJ. Propidium monoazide combined with real-time quantitative PCR to quantify viablespp. contamination in tomato products. International Journal of Food Microbiology,2013, 165(3):214-220.
[27] HONG W, XIONG J, NYARUABA R, LI J H, MUTURI E, LIU H, YU JP, YANG H, WEI H P. Rapid determination of infectious SARS-CoV-2 in PCR-positive samples by SDS-PMA assisted RT-qPCR. Science of the Total Environment, 2021, 797: 149085.
[28] Luo L X, Walters C, Bolkan H, Liu X L, Li J Q. Quantification of viable cells ofsubsp.using a DNA binding dye and a real-time PCR assay. Plant Pathology, 2008, 57(2): 332-337.
[29] Meng X L, Chai A L, Chen L, Shi Y X, Xie X W, Ma Z H, Li B J. Rapid detection and quantification of viablepv.cells in contaminated cucumber seeds using propidium monoazide and a real-time PCR assay. Canadian Journal of Plant Pathology, 2016, 38(3): 296-306.
[30] TIAN Q, FENG J J, HU J, ZHAO W J. Selective detection of viable seed-borneby real-time PCR with propidium monoazide. Scientific Reports, 2016, 6: 35457.
[31] HAN S N, JIANG N, LV Q Y, KAN Y M, HAO J J, LI J Q, LUO L X. Detection ofsubsp.in viable but nonculturable state from tomato seed using improved qPCR. PLoS One, 2018, 13(5): e0196525.
[32] Chai A l, Ben H y, Guo W t, Shi Y x, Xie X w, Li L, Li B j. Quantification of viable cells ofpv.in tomato seed using propidium monoazide and a real-time PCR assay. Plant Disease, 2020, 104(8): 2225-2232.
Establishment and Application of Rapid Quantitative Detection of Viableby PMAxx-qPCR Method
1Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing 100081;2Shandong Huasheng Agriculture Limited Company, Qingzhou 262500, Shandong
【Objective】is an obligate endoparasite that causes clubroot disease, which is the most devastating soil-borne disease in brassica crops. The propidium monoazide xx (PMAxx) could selectively bind to the chromosomal DNA of dead spores and therefore block DNA amplification by real-time fluorescent quantitative PCR (qPCR). In the present study, a strategy involving a PMAxx pre-treatment followed by the qPCR (PMAxx-qPCR) assay was developed for quantifying viable spores of,so as to provide a basis for early detection and prevention measurement of cloobroot disease. 【Method】PMA and PMAxx with concentrations of 0, 5, 10, 20, 40 and 60 µmol·L-1were prepared, respectively, and were used to pre-treatprior to DNA extraction, followed by qPCR. The inhibitory effects of PMA and PMAxx on DNA amplification ofdead spores were compared, and the optimal nucleic acid dye and concentration to distinguish between live and dead spores were determined. The illumination time was set as 0, 2, 5, 10, 15 and 20 min, respectively, and the optimal exposure time was optimized to establish a PMAxx-qPCR assay for selectively detection of viable spores of.The mixed suspensions with different ratios of dead and viable spores (0, 0.01%, 0.1%, 1%, 10%, 25%, 50%, 75% and 100% viable spores) were prepared to determine the suitability of PMAxx-qPCR assay for distinguishing viable and dead spores. The assay was also applied to quantitative detection of viable spores ofin 25 field soil samples. 【Result】PMAxx showed a better discrimination effect than PMA on the viable and dead spores of. When the concentration ofwas 1×108spores/mL, the optimal PMAxx concentration and light exposure time were 4 μmol·L-1and 10 min, respectively. The amplification of dead spores could be inhibited effectively, and only the DNA of living spores was targeted for selective amplification. For pre-defined ratio of viable spores, there was a good linear relationship between the lg of theDNA concentration assessed by PMAxx-qPCR and the theoretical viability (2=0.992). For soil samples, viablewas quantified in 11 of 25 samples, with infestation levels of approximately 32.35-6.97×103fg·g-1. 【Conclusion】The established method could quantitatively detect the viable spores of, with advantages of rapid, efficiency and sensitivity, which could be useful for avoiding the inability of qPCR method to distinguish between viable and nonviable spores. Application of the assay may potentially improvecontrol and disease management.
;clubroot disease; propidium monoazide xx (PMAxx); real-time fluorescent quantitative PCR (qPCR); viable spore
2021-11-18;
2021-12-20
国家重点研发计划(2021YFD1600303)、中国农业科学院科技创新工程(CAAS-ASTIP-IVFCAAS)、国家大宗蔬菜产业技术体系(CARS-23)、农业农村部园艺作物生物学与种质创制重点实验室开放课题(IVF2017)
李晓菁,E-mail:1041255425@qq.com。通信作者柴阿丽,E-mail:chaiali@caas.cn。通信作者李宝聚,E-mail:libaojuivf@163.com
10.3864/j.issn.0578-1752.2022.10.005
(责任编辑 岳梅)
