Ti 与Ag 离子顺次注入SiO2合成TiO2 纳米结构及其光催化活性
2022-06-28杨佳慧王刚刘昌龙
杨佳慧 王刚 刘昌龙,2
1(天津大学理学院 天津 300350)
2(天津市低维功能材料物理与制备技术重点实验室 天津 300350)
自从1972 年Fujishima 和Honda 首次利用光照实现TiO2电极分解水产氢以来[1],TiO2因其化学性质稳定、耐光、无毒、成本低等优势,一直备受关注,现已广泛应用于太阳能电池[2]、传感器[3]和光催化[4]等领域。然而,大量的研究发现,由于TiO2带隙较宽,只能吸收紫外光,并且产生的电子-空穴对极易复合,这使得TiO2的应用受到限制,需要对其改性来提升性能。目前,常用的改性方法有贵金属修饰[5]、元素掺杂[6]以及与其他半导体复合[7]等。其中元素掺杂不仅能做到拓宽光响应范围,提高光能的利用率,还能促进电子-空穴对的分离[8]。
同时,研究者也将TiO2制备成具有特殊形貌的纳米材料[9-11],用以提升TiO2的物理性能(如比表面积、吸附能力和电子传输效率等)。目前,TiO2纳米材料的合成方法主要集中在类似溶胶凝胶法[12]、水热法[13]和沉淀法[14]等的液相法。虽然此类方法制备纳米材料对仪器设备要求不高,简单经济,但其进行掺杂时,掺杂比例严重受基体固溶度的限制,而且形成的纳米结构与基底接连不够牢固,容易脱落。而离子注入技术作为一种制备纳米材料的技术和掺杂改性的方法,并不需要考虑基底材料固溶度的限制,而且制备出的纳米结构镶嵌于基底的浅表面层,比其他方法制备的纳米材料更加牢固。事实上,在2013 年,Ren等[15]就通过离子注入技术以及热处理在不同的基底材料上成功合成了TiO2,并研究了其在抑菌和光催化方面的应用。近几年,Wang 等[16]和Mu等[17]也利用Ti 和Zn 离子以及Ti 和Cu 离子顺次注入SiO2,并在退火处理后合成了棒状的TiO2纳米结构,该纳米结构的光吸收范围得到拓宽,光催化性能也有了提升,由此可见,双离子顺次注入可以有效调节纳米材料的形貌、结构和性能,在纳米材料的合成与改性中有着巨大的应用前景。
本文通过Ti离子单注入以及Ti和Ag离子顺次注入SiO2基底,经过N2气氛的退火处理,合成了TiO2纳米结构,并研究了该纳米结构的形貌、结构、光学特性和光催化性能。发现当退火温度达到700 ℃时,形成的纳米结构主要为锐钛矿相,升温到1 000 ℃时,则全部转变成金红石相。Ag 的引入使形成的纳米结构尺寸更大,并且使TiO2的初始形成温度降低。在光催化降解罗丹明B的实验中,顺次注入的样品也表现出优于单注入样品的光催化活性,同时我们对其光催化活性提升的机理进行了讨论和分析。
1 实验与表征
1.1 样品的制备
选用厚度约为1 mm的双面抛光的SiO2玻璃作为研究用的基底材料,将45 keV Ti 离子单注入或与70 keV Ag 离子顺次注入到SiO2中,Ti 和Ag 离子的注量分别为1.5×1017cm-2和3×1016cm-2。为了表述方便,Ti离子单注入及Ti与Ag离子顺次注入制备得到的样品分别命名为Ti样品和Ti+Ag样品。Ti和Ag离子的注入在北京师范大学射线束技术与材料改性重点实验室中的MEVVA II A-H型金属蒸汽真空弧(Metal vapor vacuum arc)注入机上完成,注入前,为确保SiO2的高清洁度,利用去离子水和酒精在超声波清洗机中对其进行清洗。注入过程中,为保证注入离子的均匀性,靶盘以约5 r/min 转速均匀转动。同时,为了防止离子注入导致样品温度升高,离子注入的束流密度控制在2 μA/cm2左右。离子注入完成后,将制备的样品在N2气氛下进行退火处理,温度从400~1 000 ℃,间隔温度100 ℃,保温时间为1 h。
1.2 样品的表征
利用MultiMode-8型原子力显微镜(AFM)观测了所制备样品的表面形貌,测试中设定的工作模式为敲击模式,测试范围为2 μm×2 μm。通过双光束紫外-可见-近红外分光光度计(UV-3600)对样品的光吸收进行测量,测量范围为200~800 nm。使用激光共聚焦拉曼光谱仪(LabRAM HR evolution)对样品进行拉曼光谱的表征,分析了样品结构的变化。测试中使用的激光波长为532 nm,曝光时间为1 s,累计次数为5 次,测试范围为100~1 000 cm-1。此外,还对所制备的部分样品进行了室温光致发光(PL)和X 射线光电子能谱(XPS)的表征,PL 测试是在inVia reflex 型激光拉曼光谱仪上完成的,测试时采用的激光波长为325 nm,测试范围为350~950 nm。XPS 表征则是在ESCALAB Xi+型X 射线光电子能谱仪上进行的,测试中以Al Kα为X射线源(λ≈0.834 nm)。
1.3 光催化实验
为了研究所制备的TiO2纳米颗粒的光催化性能,本文还进行了光催化实验。光催化使用的降解试剂为15 mg/L的罗丹明B(RhB)溶液,实验时,将所制备的样品切割成8 mm×8 mm 大小,然后将其放入RhB 溶液中并在黑暗环境中进行1 h 的吸附,随后进行光照实验,光照实验所使用的光源为300 W 的氙灯(300~2 500 nm),最后每隔1 h 对被降解溶液进行一次光吸收测试。
2 结果与讨论
2.1 表面形貌
图1给出了在不同温度下处理的Ti和Ti+Ag样品上测量得到的表面形貌原子力显微镜(AFM)图像。从图1(a)可以看到,对于Ti 样品,注入态时表面较为平整,随着退火温度的升高,表面逐渐有近似为球形凸起的纳米结构生成,且其尺寸随着退火温度的升高而逐渐变大。当退火温度达到900 ℃及以上时,呈现拉长形状的纳米结构逐渐增多。而对于Ti+Ag 样品(图1(b)),尽管表面形貌的热变化规律与Ti 样品相似,但在相同的热处理条件下,表面出现的纳米结构的尺寸明显更大。可见,Ag 离子的附加注入可以明显地促进表面纳米结构的热生长。

图1 经不同温度退火的Ti(a)和Ti+Ag(b)样品上测量得到的表面形貌AFM图像Fig.1 AFM images of the Ti(a)and Ti+Ag(b)samples before and after annealing at different temperatures
2.2 光吸收和带隙
图2给出了经不同温度退火后的Ti和Ti+Ag样品上测量得到的紫外-可见(UV-Vis)光吸收谱。从图2(a)可以看到,Ti 样品在可见光范围内的吸收随着退火温度升高逐渐减弱,这源于SiO2基底中注入产生的结构损伤的热恢复。经500 ℃退火后,Ti 样品在紫外波段出现明显的吸收带边,该吸收带边的出现可归因于基底中TiO2的形成[15]。结果表明:在该温度热处理后,注入的Ti 原子开始氧化,形成TiO2。同时,可以推测图1(a)中AFM 观测到的表面凸起很可能就是TiO2纳米颗粒形成引起的。进一步提高退火温度,吸收带边会发生红移,且在1 000 ℃退火后,出现了较大的红移。对于Ti+Ag 样品(图2(b)),虽然其光吸收谱的整体变化趋势与Ti 样品相似,但对应于TiO2的吸收带边在400 ℃就能出现,说明Ag 离子的附加注入可以促使TiO2在更低温度下生成。事实上,高剂量的低能重离子注入SiO2会在注入层产生大量的断键,尽管随后的热处理是在缺氧的N2气氛下进行,但注入的Ti 原子仍然可以与断键产生的氧优先结合,形成Ti的氧化物。由于Ag离子的附加注入会比Ti 离子单注入产生更多的断键,因此,热处理条件下,会有更多断键产生的氧与Ti 原子发生作用,导致Ti 原子氧化的热处理温度降低。另外,高剂量的金属离子注入SiO2,相应的金属纳米颗粒的形成在低温范围内主要通过Ostwald熟化机制热生长[18],Ag离子的附加注入会在SiO2中产生更多的缺陷,这些缺陷会增强Ti 原子的扩散,有助于Ti 原子的团聚,因而,在相同的热处理条件下会生成更大的纳米颗粒。除此之外,从图2(b)可见,对于注入态的Ti+Ag 样品,在402 nm 处还可以观测到一个弱的吸收峰,该峰对应于Ag纳米颗粒的等离子共振(SPR)吸收峰[19],说明附加的Ag 离子注入在SiO2中产生Ag的纳米颗粒,高剂量Ag离子注入SiO2导致Ag纳米颗粒形成已广泛被报道[19-21]。随着退火温度增加,该Ag SPR峰经历了增强、红移再到减弱、蓝移等系列变化,表明了Ag纳米颗粒经历了生长、氧化和最终在高温热处理下分解的过程。
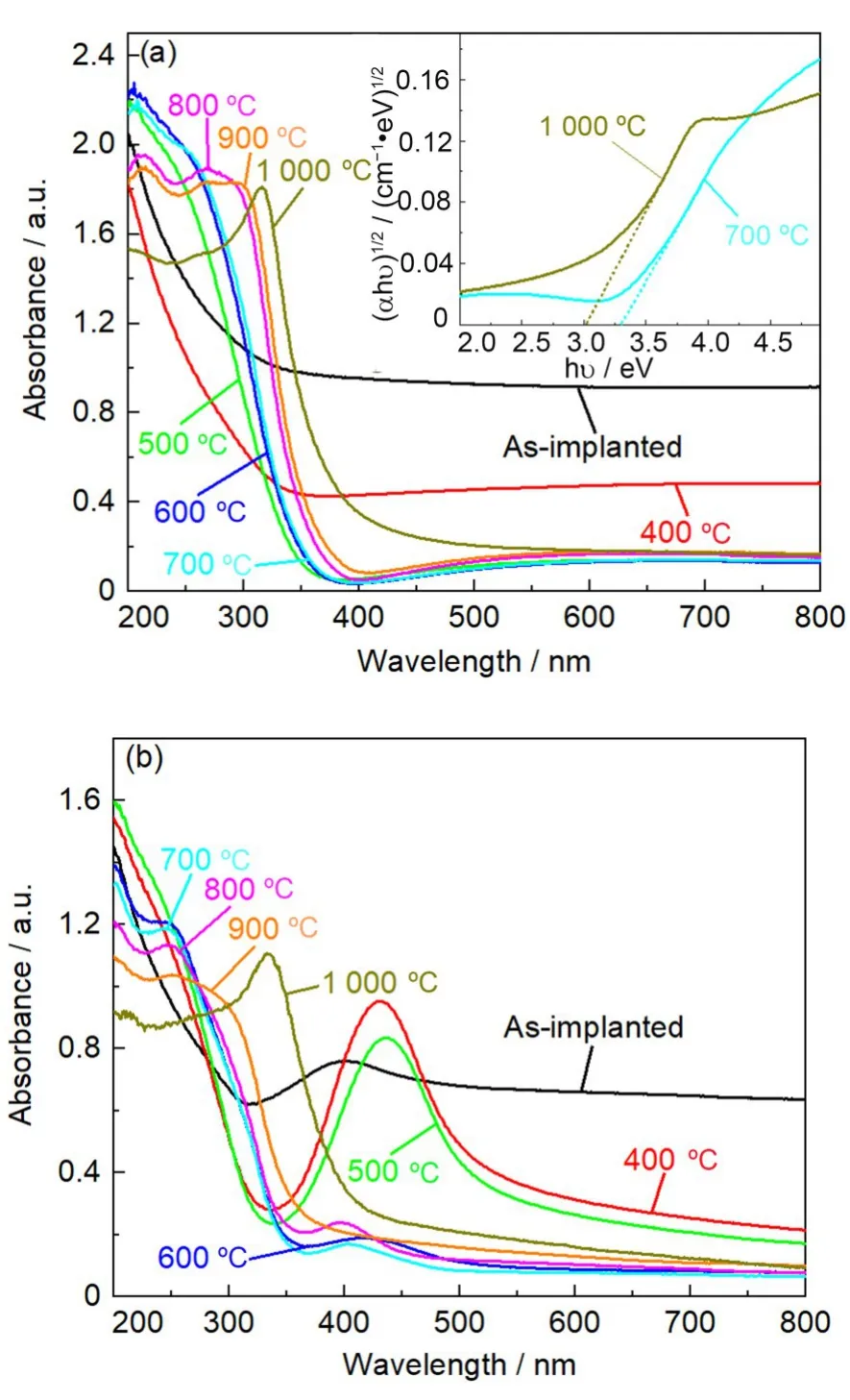
图2 在经不同温度退火后的Ti(a)和Ti+Ag(b)样品上测量得到的UV-Vis光吸收谱;图(a)给出的插图为部分Ti样品对应的Tauc曲线Fig.2 UV-Vis absorption spectra of the Ti(a)and Ti+Ag(b)before and after annealing at different temperatures;The corresponding Tauc curves from some of annealed Ti samples are also inserted in figure(a)
根据所测试得到UV-Vis 光吸收谱图,采用Tauc Plot法[22]并运用公式(1)可以来估算所形成的TiO2纳米颗粒的光学带隙。

式中:α是光吸收系数;h是普朗克常数,J·s;ν是光频率,Hz;A是常数;Eg是光学带隙,eV。指数n依赖于材料的性质,对于直接带隙半导体材料,取1/2,而对于间接带隙半导体材料,则取2。由于TiO2为间接带隙半导体,因此,这里n取2。作为例子,根据Tauc Plot 法并应用公式(1)对700 ℃和1 000 ℃退火的Ti样品的光吸收谱进行处理的结果作为例子在图2(a)的插图中给出。图中曲线的斜率直线与横轴的交点处的值就是拟合得到的光学带隙值。通过Tauc Plot 法处理得到的各热处理温度下Ti 和Ti+Ag 样品中所形成的TiO2纳米颗粒的光学带隙值列于表1中。从表1可以清楚地看到,随退火温度增加,无论是Ti样品还是Ti+Ag 样品,所形成的TiO2纳米颗粒的光学带隙逐渐减小,尤其是Ti+Ag样品在高温退火下不仅其光学带隙相较于Ti 样品更小,而且减小的幅度更大。已有实验结果表明[23-25],由于量子尺寸效应,纳米颗粒的光学带隙会随其尺寸减小而增大,并且TiO2纳米颗粒尺寸在30 nm以下时,有较强的量子尺寸效应,在30 nm以上时,尺寸变化对光学带隙的影响减小,其光学带隙几乎等同于块体材料的光学带隙。从图1可知,随着退火温度的升高,纳米颗粒的尺寸逐渐变大,这也是光学带隙逐渐变小的原因,但在700 ℃及更高温度退火后,纳米颗粒的尺寸超过30 nm,光学带隙的变化幅度反而增大,我们认为这应该与TiO2纳米颗粒在热处理过程中由锐钛矿相(光学带隙约3.2 eV)向金红石相(光学带隙约3.0 eV)的转变密切相关,这将通过下面的Raman测试得到证实。
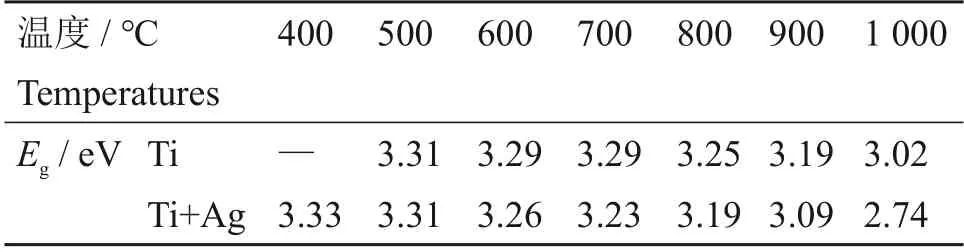
表1 Ti和Ti+Ag样品在不同温度退火后拟合得到的光学带隙结果Table 1 Calculation results of optical band gap of the Ti and Ti+Ag samples after annealing at different temperatures
2.3 Raman光谱与相变
图3给出了在不同温度退火后的Ti和Ti+Ag样品上测量得到的Raman 光谱图,为了对比,图中还给出了未经注入的纯SiO2基底上测量得到的Raman谱图。从图3可以看到,离子注入会导致纯SiO2的Raman 特征峰消失,但随后的热处理会使得基底材料注入损伤逐渐恢复,相应的特征峰也随之逐渐出现。对于Ti 样品(图3(a)),当退火温度达到700 ℃时,在143 cm-1左右出现了一个新的特征峰,对应于锐钛矿相TiO2的特征峰[26],表明在700 ℃时形成的主要是锐钛矿相的TiO2。随着退火温度的进一步升高,该特征峰变弱,并在900 ℃退火后消失,同时在128 cm-1、208 cm-1、235 cm-1、462 cm-1和614 cm-1左右出现了新的特征拉曼峰,且在446 cm-1出现肩。其中,235 cm-1、446 cm-1和614 cm-1为金红石型TiO2的特征峰[27],而128 cm-1、208 cm-1和462 cm-1则为SiO2结晶相的特征峰[28]。以上结果表明,进一步升高退火温度会导致锐钛矿型TiO2逐渐转变为金红石型TiO2,同时伴随着SiO2的相变,在基底中出现了SiO2的结晶相。1 000 ℃退火后,金红石相和SiO2结晶相的峰进一步增强,说明两者含量增加。通常来说,无定型的SiO2在论文所涉及的热处理温度范围内难以形成结晶相,我们认为,之所以在离子注入且经高温退火的SiO2中出现了SiO2的结晶相,其原因可能跟SiO2中合成的TiO2有关。事实上,Okabayashi等[29]在无定型SiO2中掺杂TiO2和CaO,发现经高温热处理后,会出现结晶相的SiO2,他们认为掺入的金属氧化物会在SiO2结晶过程中起到催化剂的作用。
如图3(b)所示,对于Ti+Ag 样品,其Raman谱随退火温度变化的规律大致与Ti 样品类似。不过,相比于Ti样品,Ti+Ag样品经500 ℃退火后就出现了锐钛矿相TiO2的特征峰,说明在500 ℃退火后就形成了Raman 技术可探测的锐钛矿相TiO2,这与以上AFM和UV-Vis测试结果是一致的,表明附加Ag 离子的注入不仅能够降低TiO2的形成温度,而且能促进TiO2的热生长。

图3 不同温度热处理的Ti(a)和Ti+Ag(b)样品测量得到的Raman谱图Fig.3 Raman spectra of the Ti(a)and Ti+Ag(b)samples before and after annealing at different temperatures
以上Raman 结果清楚地表明,无论是Ti 样品还Ti+Ag样品,低温热处理(700 ℃及以下温度)在SiO2基底表面形成的主要是锐钛矿相的TiO2,继续升高退火温度,锐钛矿相的TiO2逐渐向金红石相TiO2转变,且在1 000 ℃时几乎全部转变成金红石相TiO2。因此,表1中通过光吸收谱拟合得到的光学带隙值变化也正是TiO2发生相变导致的。至于Ag 离子的附加注入使得所合成的TiO2的光学带隙在高温下明显比单Ti 离子注入的情形要低,其原因可能在高温下Ag原子使TiO2中形成了更多的氧空位[30]。
2.4 光催化活性与催化机理分析
为探究所合成的TiO2的光催化性能,我们选用了经700 ℃和1 000 ℃退火的Ti 和Ti+Ag 样品来光催化降解RhB,两个温度退火下合成的TiO2分别对应于锐钛矿相和金红石相,实验得到的结果如图4所示。从图4可以看到,无论形成的是锐钛矿相还是金红石相的TiO2,Ti+Ag的样品降解RhB的效果均高于Ti 样品。与Ti 样品相比,700 ℃和1 000 ℃退火下的Ti+Ag 样品的降解活性可以分别提升13.68% 和23.96%,同时,Ti+Ag 样品1 000 ℃退火后的降解率也比700 ℃退火后提升了9.36%。可见,Ag离子附加注入能有效提高所合成的TiO2纳米颗粒的光催化活性,且对1 000 ℃退火后的金红石相的提升效果更强。
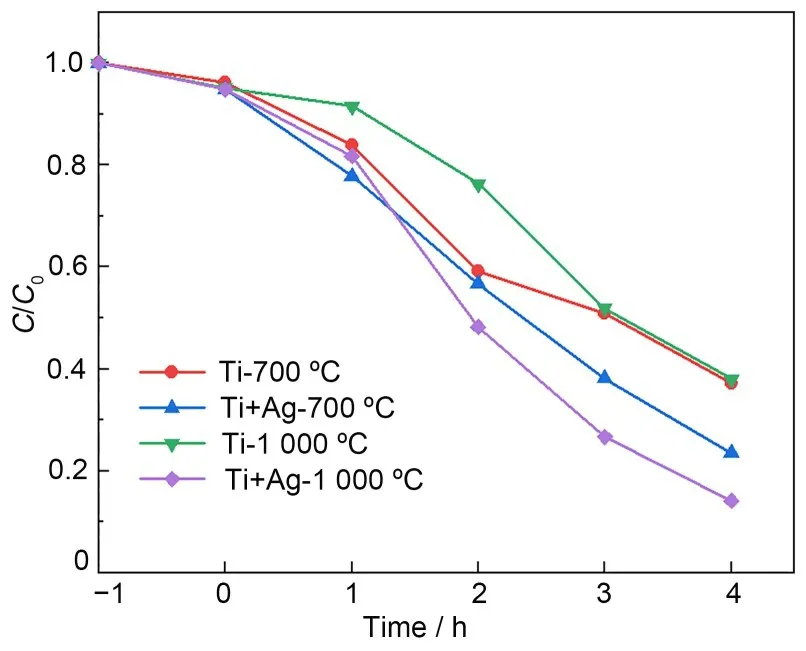
图4 经700 ℃和1 000 ℃退火后的Ti和Ti+Ag样品对RhB的光催化降解曲线Fig.4 Photocatalytic degradation curves of RhB obtained in the Ti and Ti+Ag samples after annealing at 700 and 1 000 ℃,respectively
无论是锐钛矿相还是金红石相的TiO2,都是常用的光催化剂,其催化活性依赖于多种因素,如比表面积、孔径大小和颗粒尺寸等,此外,制备方法也会影响其光催化活性[31-33]。但通常锐钛矿相的光催化活性强于金红石相,从实验结果来看,对于Ti 样品,锐钛矿相的光催化活性更高,与文献[34]报道一致;而对于Ti+Ag 样品,Ag 离子的附加注入使锐钛矿相和金红石相的光催化活性都得到了提升,并且使金红石相的光催化活性超过锐钛矿相。
为了探究Ag 离子附加注入提升所合成的TiO2纳米颗粒的光催化活性的原因,我们针对700 ℃和1 000 ℃退火的Ti 和Ti+Ag 样品进行了PL 和XPS的测试,详细研究了所合成的TiO2纳米结构的发光特性及其元素分布和价态,测试得到的结果如图5、图6所示。

图5 经700 ℃(a)和1 000 ℃(b)退火后的Ti和Ti+Ag样品上测量得到的PL谱图,其中图(b)中插图为400 nm到650 nm的放大图Fig.5 PL spectra of the Ti and Ti+Ag samples after annealing at 700 ℃(a)and 1000 ℃(b),respectively.Inserted in figure(b)shows the magnified curves from 400 nm to 650 nm
从图5(a)可以看到,对于700 ℃退火后的Ti和Ti+Ag 样品,其最强发光均出现在近红外区域。Knorr 等[35]曾报道,对于TiO2来说,少量的金红石相的存在则会引起很强的近红外发光。因此,尽管Raman测试(图3)表明,无论是Ti还是Ti+Ag样品,经700 ℃退火形成的TiO2纳米颗粒主要以锐钛矿相存在,但通过PL 测试分析显示,所形成的TiO2纳米颗粒中应该已有少量锐钛矿相转变成金红石相。另外,从图5(a)还可以发现,Ti+Ag样品的发光强度稍弱于Ti 样品,表明Ti+Ag 中电子-空穴对的复合效率有所下降。对于1 000 ℃退火的Ti和Ti+Ag 样品(图5(b)),它们在820 nm 处均出现了强的近红外发光,该波长处出现的发光可以归因于金红石相TiO2的形成[35],具体而言,它跟金红石型TiO2中的间隙氧密切相关[17]。与Ti样品相比,Ti+Ag 样品在820 nm 处发光的强度明显低于Ti 样品,不仅表明了其内部合成的金红石型TiO2的电子-空穴对复合被抑制,而且TiO2结构中的间隙氧的含量明显降低。除此之外,对于1 000 ℃退火的Ti 和Ti+Ag 样品,还在575 nm 左右处观测到一个弱的发光带,该发光带经放大处理后作为插图示于图5(b)中,该发光带来源于TiO2的本征缺陷氧空位[36]。从该插图可以看到,Ti+Ag 样品上的该发光带其强度高于Ti 样品,意味着所形成的TiO2纳米结构中含有更多的氧空位。因此,对于Ti+Ag的样品来说,热处理无论是引起TiO2纳米结构中间隙氧的减少,还是其氧空位的增加,都说明该结构中氧的含量减少。氧含量减少的原因可能与Ag的引入有关。一方面,Ag离子的附加注入可以大大地促进TiO2纳米结构的热生长,这会导致有更多的氧参与Ti 原子的氧化和生长;另一方面,在热处理升温过程中,注入的Ag原子也会发生氧化,生成Ag的氧化物,消耗掉一部分氧,但氧化银并不稳定,在高温下中会再次分解并熔解析出。
700 ℃和1 000 ℃退火后的Ti 和Ti+Ag 样品上分析得到的XPS 谱图(图6)。从图6(a)可以看到,两类样品表面主要存在C、O、Si、Ti 四种元素,其中,C来源于表面的污染吸附,此外,700 ℃退火的Ti+Ag 样品上还测得微弱的Ag 信号,表明还保留了少量的Ag,这与UV-Vis光吸收测试结果是一致的,如图2(b)所示,在该温度下,尽管Ag SPR峰很弱,但依然清晰可见。

图6 Ti和Ti+Ag样品在700 ℃和1 000 ℃退火后的(a)XPS全谱和(b)Ti2p、(c)O1s、(d)Si2p、(e)Ag3d的XPS精细谱Fig.6 (a)XPS survey spectra and high-resolution XPS spectra of(b)Ti2p,(c)O1s,(d)Si2p,and(e)Ag3d of Ti and Ti+Ag samples annealed at 700 ℃and 1 000 ℃

从图6(b)看到,Ti样品无论是700 ℃退火后,还是1 000 ℃退火后,都在458.15 eV 和463.90 eV附近出现特征峰,分别对应于TiO2中的Ti4+2p3/2和Ti4+2p1/2[37];而对于Ti+Ag 样品,700 ℃退火后Ti2p 峰位向高能方向移动,移动至458.31 eV 和464.02 eV,这是由于样品中Ag 的存在,Ag 的电负性大于Ti,使得Ti 对电子的吸引变弱,从而降低Ti的核外电子密度,电子结合能变大。Ti+Ag样品1 000 ℃退火后由于Ag 已经熔解析出,Ti2p 的峰位又回到458.14 eV和463.93 eV结合能处。对Ti和Ti+Ag样品的O1s精细谱进行拟合,如图6(c)所示。可以看到,700 ℃退火的Ti 样品在529.49 eV和531.55 eV 结合能处有两个峰,分别对应于TiO2中的O[37]和SiO2中的O[38];而经1 000 ℃退火后,除了对应于TiO2中的O的峰(529.47 eV)外,拟合结果还显示,Ti样品在530.90 eV和532.22 eV处还出现了两个峰,分别对应于Ti-O-Si和表面吸附羟基中的O[38-39]。对于700 ℃和1 000 ℃退火的Ti+Ag样品,均在529.5 eV、530.9 eV 和532.2 eV 左右处出现O的峰,同样分别对应于TiO2、Ti-O-Si和表面吸附羟基中的O。不过,与700 ℃退火的样品相比,1 000 ℃退火下,Ti+Ag 样品中表面吸附羟基氧含量最多,它在三类氧中的占比高达78.9%,表明吸附羟基的能力显著增强。两类样品中Ti-OSi 的产生可能与Ti 的热氧化有关,在Ti 原子与断键产生的氧结合形成氧化物过程中,可能存在部分氧并没有与Si完全断开键合,因而产生了Ti-OSi结构。同时,正因为Ti-O-Si的存在,导致了材料的亲水性增强[40],从而使得Ti-O-Si 中O 和表面吸附羟基中的O 同时出现。从图6(d)中Si2p 的精细谱可以看到,Ti样品在700 ℃退火后,峰出现在约102.26 eV,对应于SiO2中的Si[38]。而在1 000 ℃退火后,该峰位移至102.95 eV,对应Ti-O-Si 中的Si[38],同样证实了Ti-O-Si 的产生。对于Ti+Ag 的样品,无论是700 ℃退火还是1 000 ℃退火,XPS 分析到的Si2p 的峰位其结合能均有所增大,分别为102.77 eV和103.16 eV,这说明,在样品中都生成了Ti-O-Si。
除此之外,通过对退火的Ti+Ag 的样品分析,我们在700 ℃退火后的样品中检测到Ag 元素的存在,如图6(e)所示。位于367.49 和373.58 eV 的两个峰分别对应于Ag 3d5/2和Ag 3d3/2[41],说明Ag 元素以Ag0的形式存在。而1 000 ℃退火后并没有分析到Ag的特征峰,表明高温退火后Ag几乎全部从样品中析出了,这与UV-Vis光吸收谱中Ag的SPR峰的变化得出的规律是一致的。
在光催化反应中,当能量大于半导体带隙的光子入射到半导体上时,价带上的电子会跃迁到更高的能级,这样就产生了电子-空穴对。电子和空穴会进一步作用于吸附在半导体表面的O2、OH-、H2O 等产生强氧化还原能力的超氧自由基(·O2-)和羟基自由基(·OH),这些活性物质则再进一步降解有机污染物。而·O2-和·OH的产生不仅依赖于电子-空穴对的分离,更依赖于半导体导带与价带的电位,我们可以通过n型半导体的导带计算经验公式(2)[42]对半导体的导带电位进行估算。

式中:E是导带的电位;X是绝对电负性;Ec是氢尺度上的自由电子的能量(~4.5 eV)、Eg是半导体材料的带隙,并且以上变量单位都为eV。对于Ti+Ag 700 ℃退火后的样品,我们将TiO2的绝对电负性5.81 eV和计算出的带隙3.23 eV代入,则得到导带电位约为-0.305 eV,进一步计算出价带电位约为2.925 eV。导带电位比O2/·O2-的氧化还原电位(-0.28 eVvsNHE)[43]偏负,则导带上的光生电子与O2反应可形成强氧化性的·O2-。同时,价带电位比·OH/OH-和·OH/H2O 的氧化还原电位(+1.99 eV和+2.27 eVvsNHE)[43]偏正,价带上的空穴也能将H2O和OH-转化成·OH。同样,我们也可以计算出Ti+Ag样品1 000 ℃退火后的导带和价带的电位分别为-0.06 eV 和2.68 eV。可见,导带上的光生电子不能还原O2,但价带上的光生空穴则可以将H2O和OH-氧化成·OH。
对于700 ℃退火后的Ti+Ag样品,其光催化活性相比于Ti 样品有所提升,其原因可能跟以下几方面有关。首先,从AFM 的测试结果可知,Ti+Ag样品表面形成了比Ti样品更大的纳米颗粒,在相同的测试投影面积(2 μm×2 μm)下,AFM 测试给出的Ti 样品中纳米颗粒的表面积约为4.01 μm2,而Ti+Ag 样品中相应的表面积约为4.30 μm2。TiO2纳米颗粒表面积的增加,可以使样品与更多的溶液接触,加快催化反应进程。其次,由于在Ti+Ag样品中有Ag的存在,它可以与TiO2接触形成了肖特基结[44],经光照后,价带上的电子跃迁到更高的能级,形成电子-空穴对。由于TiO2的费米能级高于Ag,跃迁到导带上的电子会在费米能级高度差的作用下转移到Ag上,与此同时,空穴就停留在了TiO2的价带上,实现了电子与空穴的分离。事实上,PL 也证实了Ti+Ag 样品比Ti 样品具有更低的电子空穴复合率。此外,XPS测试结果显示,在Ti+Ag 样品中还形成了Ti-O-Si 亲水结构以及出现表面吸附羟基氧,这也会产生更多强氧化能力的·OH。
而Ti+Ag样品在1 000 ℃退火后,Ag已经全部熔解析出,因而其光催化活性提升的原因与700 ℃退火后的样品并不相同。首先,如UV-Vis 光吸收谱所示(图2(a)),其吸收带边红移到可见光波段,这使得光催化反应可以利用到更多的光能,进而产生更多的电子-空穴对。其次,PL 谱表明在Ti+Ag 样品中存在大量的氧空位,氧空位的存在既可以充当吸附反应物的活性位点,又可以做为电子的捕获陷阱,进而使反应物和电子直接接触反应,同时对电子的捕获也实现了电子-空穴对的分离[45]。最后,我们同样可以从XPS 谱中看到,样品中Ti-O-Si的形成以及样品表面更强的羟基吸附能力,这使得催化过程中可以产生更加充足的·OH。值得一提的是,尽管Ti+Ag样品在1 000 ℃退火后形成了更大的纳米结构,然而其表面积并未增加,AFM 测试结果表明,其表面积约为4.35 μm2,相比1 000℃退火后的Ti样品中的纳米结构的表面积(~4.63 μm2)稍有减小,因此,纳米颗粒表面积的变化并非是影响其催化活性的主要因素。
3 结论
通过Ti 离子单注入或Ti 和Ag 离子顺次注入,结合后续N2气氛下的热退火处理,在SiO2浅表面中合成了TiO2纳米结构,该纳米结构在700 ℃及以下退火温度主要为锐钛矿相,升高退火温度到1 000 ℃可导致其转变为金红石相。Ag离子的附加注入不仅可以降低TiO2纳米结构的形成温度和光学带隙,而且可以大大地促进TiO2纳米结构的热生长。在光催化降解罗丹明B的实验结果揭示,在Ag 离子的附加注入下,热处理形成的TiO2纳米结构的光催化活性可得到明显的提升。对于700 ℃退火形成的锐钛矿相TiO2纳米结构,注入的Ag会与TiO2接触形成肖特基结,光生电子可通过从TiO2到Ag 的转移实现了电子空穴的分离,从而使电子和空穴的氧化还原能力表现出来,提高了TiO2的催化活性。而对于1 000 ℃退火形成的TiO2纳米结构,Ti+Ag的样品中氧空位提供了吸附反应物的活性位点和捕获电子的陷阱,不仅实现了光生电子与反应物的直接接触,也实现了电子和空穴的分离,进而提升样品的光催化降解能力。同时以上发现为制备高效的TiO2催化剂提供了参考。
作者贡献说明杨佳慧负责设计和进行实验,采集整理和分析数据,以及撰写论文;王刚参与部分实验并进行指导;刘昌龙提出研究思路,进行专业指导,以及修改论文。全体作者均已阅读并同意最终的文本。
