运动通过调节自噬途径防治非酒精性脂肪性肝病研究进展
2022-06-26马穰桂夏志尚画雨
马穰桂 夏志 尚画雨
1成都体育学院运动医学与健康学院(成都 610041)
2温州大学体育与健康学院(浙江温州 325035)
3井冈山大学体育学院(江西吉安 343009)
非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)是一种以肝细胞脂肪变性和肝脏脂质沉积为特征的代谢性疾病,随病程的发展可表现为单纯性脂肪变性、非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH)、脂肪性肝纤维化,甚至发展为肝硬化和肝细胞性肝癌。NAFLD被视为代谢综合征在肝脏的表征,且与肥胖、Ⅱ型糖尿病、高血压、高脂血症、动脉粥样硬化等疾病的高发病率密切相关[1]。近20年来,我国NAFLD的患病率为13%~43%,年均发病率约4%[2],且呈现出年轻化趋势,在肥胖儿童中患病率可达68.2%[1]。鉴于此,提高对该病的认知,并在科学防治的基础上增强管理具有重要意义。
自噬包括巨自噬、微自噬以及分子伴侣介导的自噬,能清除受损或过多的细胞器(如线粒体)和降解异常蛋白质,对于维持细胞正常生理功能具有重要作用。研究表明,NAFLD患者肝细胞内通常呈现自噬受损[3],病程中自噬流(autophagic flux)阻断将进一步加重肝脏脂肪变性[4],且自噬流的阻断程度与肝损伤及炎症水平呈正相关[3]。然而,对高脂膳食诱导的NAFLD小鼠给予雷帕霉素(rapamycin)激活自噬后,肝细胞脂肪变性及损伤程度明显减轻[5]。以上研究结果均提示自噬参与了NAFLD的发生与发展。目前,适度运动与饮食热量限制仍是防治NAFLD的首要策略[1]。其中,运动不仅可通过诱导细胞自噬(autophagy)[6]、线粒体自噬(mitophagy)[7]改善脂质沉积,还能减轻胰岛素抵抗[8]、氧化应激和炎症反应[9],是防治NAFLD的重要手段。本文应用计算机检索Pubmed和Web of Science数据库及中国期刊全文数据库2001年1月至2022年3月刊发的运动干预NAFLD相关文献(中文检索词为“运动、非酒精性脂肪性肝病”,英文检索词为“exercise,nonalcoholic fatty liver disease”),梳理并归纳运动防治NAFLD过程中的肝细胞(线粒体)自噬变化,为从自噬调控角度探寻防治NAFLD的临床干预手段提供参考。
1 NAFLD的发病机制学说
目前,“二次打击”学说作为NAFLD的经典发病机制,被学界广泛接受:静坐少动的生活方式、高糖高脂饮食、肥胖和胰岛素抵抗引起肝脏脂质积累被认为是“首次打击”,致使肝脏对进一步的损伤更加敏感;随后,氧化应激增高、炎性级联反应激活以及线粒体功能异常等“二次打击”会导致炎症(NASH)和纤维化的发生[10]。另有报道指出,在“二次打击”之后还存在“三次打击”:肝脏中的修复/再生反应不足以抵抗过度氧化应激引发的肝细胞死亡,最终导致肝硬化和肝细胞性肝癌[11]。还有学者认为,无论是“二次打击”还是“三次打击”均不足以解释NAFLD进程中所有的分子和代谢变化,进而提出“多重打击”假说,认为NAFLD的发生是胰岛素抵抗、脂肪组织分泌的激素、营养、肠道微生物群、遗传和表观遗传等多种因素共同作用的结果[10],这可能为NAFLD的发病机制提供了更全面的解释。
此外,近年来研究发现自噬参与NAFLD的发病与进展:在NAFLD初期,由于脂质摄入量增多,自噬激活可降解肝细胞内过多的脂质,以减少肝内脂质蓄积,维持肝脏脂代谢稳态;然而,脂质持续大量摄入(如长期高脂饮食)将导致肝细胞自噬功能受损[12]。自噬受损则会加重肝脏脂质沉积、氧化应激和内质网应激,致使损伤线粒体清除受阻并诱发炎症反应等,加剧NAFLD的进程[13]。值得注意的是,Pi等[6]的研究表明长期规律运动能通过提高自噬通量维持脂质稳态,从而改善NAFLD相关的肝脂肪变性。鉴于自噬在NAFLD发生发展过程中所起的重要作用,深入研究运动等因素诱导的细胞(线粒体)自噬在NAFLD发病中的作用及其作用机制对于NAFLD的预防、治疗和管理意义重大。
2 NAFLD相关的自噬功能障碍
2.1 细胞自噬
细胞自噬能通过清除蛋白聚集体、受损细胞器和脂滴调节能量平衡和细胞质量控制,从而起到维持细胞内稳态的保护作用;自噬水平降低时,细胞在面对有害刺激时对死亡更为敏感。Mei等[14]最初提出细胞自噬可通过介导肝细胞内脂解抑制肝脂肪变性,从而参与NAFLD的发生进展。后续研究表明,细胞自噬下调亦与NAFLD多重进展机制(包括胰岛素抵抗、过量的甘油三酯和游离脂肪酸、内质网应激和氧化应激等)密切相关[15]。不少报道指出,包括自噬相关基因7(au-tophagy-related gene 7,ATG7)[16]、ATG14[17]或转录因子EB(transcription factor EB,TFEB)[18]缺失的肝细胞,或高脂饮食喂养的小鼠在施加自噬抑制剂3-甲基腺嘌呤处理后[19]均呈现出NAFLD加重的特征,包含泛素化p62/sequestosome 1的脂滴积聚增多,产生促炎细胞因子。由此说明,自噬可能是NAFLD发病的关键靶点,在NAFLD的发生[20]、发展到NASH[13]等关键进程中发挥多重作用。
2.1.1 AMP活化蛋白激酶(AMP-activated protein ki-nase,AMPK)
目前研究证实,能量开关因子AMPK特异性激活能通过不同机制(包括自噬激活)延缓肥胖状态下肝脂肪变性的进程[21]。二甲双胍(metformin)[22]或海藻糖(trehalose)[23]可靶向激活AMPK从而显示出良好的抗脂肪生成作用。其中,AMPK主要通过抑制下游雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)和激活UNC-51样激酶1(unc-51 like autophagy activating kinase 1,ULK1)信号途径而参与调节NAFLD条件下受损的细胞自噬[24]。例如,分别对肥胖和/或糖尿病大鼠实施减脂手术后均观察到肝脏AMPK磷酸化表达增加,随即抑制mTORC1活化,进而明显改善肝脂肪变性[25,26]。又如,AMPK能直接磷酸化自噬起始因子ULK1和Beclin-1/ATG6而促进自噬激活,然而脂肪酸转位酶CD36(主要参与肝脏脂肪酸摄取)可通过下调该途径以抑制脂肪肝中细胞自噬的启动[27]。此外,He等[28]报道指出,高脂饮食的小鼠肝内过氧化物酶体β-氧化生成的乙酰辅酶A增加会促进mTORC1定位于溶酶体表面,随后活化的mTORC1通过磷酸化ULK1而抑制细胞自噬。因此,AMPK和mTORC1是目前已知的通过激活/抑制ULK1复合物启动自噬的关键调节蛋白。
此外,AMPK和mTORC1亦可在溶酶体膜处激活进而参与调控溶酶体功能,故被视为自噬末期的重要调节因子[29]。其中,mTORC1的溶酶体定位是其激活的前提条件,故诱导mTORC1定位至溶酶体是调控其活性的主要方式之一[30]。Napolitano等[31]指出,在营养丰富的条件下,促进自噬和溶酶体生成的主要转录因子TFEB可与溶酶体膜定位的mTORC1相互作用,mTORC1介导TFEB磷酸化使其与胞浆定位的14-3-3蛋白结合,最终TFEB滞留于胞浆而丧失活性;相反,在营养剥夺或溶酶体功能障碍导致溶酶体衍生氨基酸减少的条件下,mTORC1活性被抑制,促使TFEB去磷酸化并向胞核转移,进而促进溶酶体生成与自噬激活。需要注意的是,由于mTORC1对溶酶体功能的依赖性,TFEB活化后亦可通过提高溶酶体活性而激活mTORC1,但mTORC1活化将再次通过磷酸化TFEB而抑制其发生核转位[32]。该反馈信号在肝组织中循环往复,并与脂肪变性的发展直接相关,其中诱导mTORC1激活的条件受阻(如溶酶体功能障碍)会加剧NAFLD的进程[32]。基于TFEB在通过参与细胞自噬调节脂肪变性中的重要作用,TFEB激动剂最近备受关注。例如,Wang等观察到,施加TFEB激活剂地高辛(digox-in)、斑鸠霉素(ikarugamycin)和阿来西定(alexidine)干预可有效改善高脂膳食诱导的小鼠肝脂肪变性和胰岛素抵抗,且这些变化均与肝细胞内自噬通量上调有关[33];反之,使用自噬诱导剂MSL亦能通过诱导核TFEB易位获得相似的健康收益[34]。又如,使用依折麦布(ezetimibe,一种胆固醇吸收抑制剂)[35]和海藻糖[36]可分别在肝细胞和巨噬细胞内激活TFEB而诱导自噬,并在巨噬细胞内抑制NOD样受体蛋白3依赖的白介素-1β生成,最终依折麦布有效减轻了NAFLD小鼠的肝损伤及脂肪变性,而海藻糖干预则显示出对动脉粥样硬化的保护作用,但TFEB激活剂能否有效抑制与NASH进展相关的炎症反应尚不清楚。目前已有学者开展依折麦布干预NASH患者的临床试验,结果显示6个月的依折麦布治疗能显著下调NASH患者血清转氨酶和血脂水平以及肝脂肪变性程度,但并未明显改善肝纤维化[37],未来还需更大规模的实验进一步研究TFEB激活剂对NAFLD的防治效益。除了与TFEB互相作用之外,mTORC1激活还能通过介导自噬调控蛋白Pacer(pro-tein associated with UVRAG as autophagy enhancer)失活而抑制NAFLD小鼠肝细胞内自噬体与溶酶体相融合(fusion autophagosomes with lysosomes)[38]。综上,溶酶体定位的mTORC1可通过与TFEB、Pacer相互作用以调节溶酶体功能,进而在细胞自噬后期发挥关键作用。如前所述,mTORC1的活化状态亦可被上游的AMPK调控。尽管研究表明AMPK可通过调节不同生理或病理条件下TFEB的激活、溶酶体膜V-ATP酶活性和钙离子(Ca2+)转运而影响溶酶体功能[39],但目前类似机制在NAFLD模型中的研究尚有待进一步深入探讨。
另外,AMPK还能通过激活去乙酰化酶1(Sirtuin 1,SIRT1)而参与调节NAFLD条件下受损的肝细胞自噬。Wang等[40]报道指出,在给予18周高脂饮食喂养的小鼠肝脏中或棕榈酸处理的HepG2肝细胞内均呈现出AMPK磷酸化水平和SIRT1蛋白表达明显下调且自噬受阻,使用siRNA干扰SIRT1表达会进一步抑制自噬。此外,在激活和/或抑制AMPK后SIRT1蛋白表达及自噬水平升高和/或下降,提示AMPK/SIRT1通路介导NAFLD状态下的肝细胞自噬。相似的是,Tong等[41]的研究亦在20周高脂饮食喂养的小鼠肝内观察到SIRT1、SIRT3、叉头框蛋白O3a(forkhead box protein O3a,FOXO3a)蛋白表达和自噬水平均显著下调,施加4周利拉鲁肽干预能逆转上述结果,最终激活自噬以改善NAFLD。鉴于SIRT1/3位于FOXO3a上游[42,43],上述研究结果提示AMPK-SIRT1/SIRT3-FOXO3a通路可能在NAFLD致肝细胞自噬受损中发挥重要作用。
2.1.2 其他细胞自噬相关因子
总体上,NAFLD病理条件下细胞自噬普遍减弱[3]。肝细胞中多余的游离脂肪酸和葡萄糖转化为甘油三酯后储存在脂滴中,脂滴的形成需要微管相关蛋白1轻 链3(microtubule-associated protein 1 light chain 3,LC3)与ATG7的参与[44];ATG7缺失会致使自噬受损,进而诱导内质网应激并加重胰岛素抵抗[45],以上结果表明细胞自噬与脂质代谢、胰岛素抵抗和肝细胞损伤有关。然而,关于细胞自噬途径的某些相关因子在脂肪变性中的作用仍存在争议:例如,肝细胞特异性敲低ATG3会减轻肝脂肪变性[46],而特异性敲除ATG7则可能加重[47]或减弱[48]肝脂肪变性;肝细胞缺失启动自噬体形成的ULK1复合物核心蛋白FIP200(focal adhe-sion family kinase-interacting protein of 200 kDa)也可减少高脂膳食小鼠肝脏甘油三酯的含量[49]。尽管上述实验结果有待进一步研究确认,但在NAFLD在体和离体模型中,自噬激活剂的使用仍显示出积极的效果。例如,塞来昔布等化合物可通过修复自噬通量而缓解NAFLD[50]。又如,具有促自噬特性的锌等矿物质,也被证明可促进肝脂肪自噬并减少肝脂肪变性[51]。再如,给予卡马西平和雷帕霉素干预亦可减轻高脂饮食诱导的NAFLD小鼠肝自噬受损及脂肪变性,同时有效增强胰岛素敏感性[5]。
此外,长期脂质堆积可引起多种组织中Ca2+信号异常改变,加剧肥胖状态下的代谢失调[52]。在脂肪变性的肝细胞内观察到胞浆Ca2+浓度升高,而内质网Ca2+水平和钙池操纵性钙内流的活性降低,这些变化加速了NAFLD的进展[53]。基于此,对肥胖小鼠给予钙通道阻滞剂维拉帕米处理,可明显改善肝细胞胞浆内异常升高的Ca2+水平,同时受阻的自噬体与溶酶体融合过程亦得以恢复[54]。另有报道指出,在脂肪变性的肝内ATG7蛋白表达下降与钙蛋白酶的蛋白水解活性升高有关[45]。上述研究结果表明,Ca2+信号的改变会影响NAFLD进程中多重自噬调控步骤,是细胞自噬活性的重要调节因素,但其调控机制有待进一步阐明。
2.2 线粒体自噬
线粒体为响应代谢需求而发生形态和功能变化。在6周蛋氨酸胆碱缺乏饮食诱导的NASH小鼠模型中,观察到肝内线粒体肿胀和自噬中间产物积聚,然而肝脏特异性敲除线粒体融合基因OPA1能明显逆转上述线粒体异常现象并有效减轻肝损伤,表明线粒体动力学和自噬在代谢适应营养物质输入过程中发挥重要作用[20]。
通过线粒体自噬清除受损的线粒体被视为可以延缓NAFLD进程的一种保护机制。不少报道指出,在高脂饮食诱导的动物模型以及施加油酸或棕榈酸处理的离体细胞中,均可观察到线粒体自噬受阻,且该现象与一系列NAFLD相关表征(包括脂肪积聚增加、炎症反应和氧化应激加剧)有关[55,56]。现已证实,NAFLD状态下存在PTEN诱导激酶1(PTEN-induced putative ki-nase protein 1,PINK1)/Parkin(Parkinson protein 2)[55-57]和Bcl-2/腺病毒E1B相互作用蛋白3(BCL2/adeno-virus E1B protein-interacting protein 3,Bnip3)[58]等信号途径参与介导线粒体自噬,在适宜的调控作用下则可能修复肝细胞内线粒体自噬功能,并最终改善代谢结局。
2.2.1 PINK 1/Parkin与NAFLD
PINK1和Parkin是两个帕金森症相关蛋白,其中PINK1位于Parkin的上游,二者协同介导了受损线粒体表面结构或功能蛋白的多聚泛素化过程,在去极化线粒体自噬降解中发挥关键作用[56]。研究发现,与野生型小鼠呈现出正常线粒体结构相比,Parkin基因敲除小鼠在摄入酒精后,其肝脏线粒体氧化应激损伤明显加重,呈现出线粒体肿胀且嵴结构缺失[59]。上述现象在高脂饮食诱导的NAFLD模型小鼠中亦得到证实,该模型下肝脏PINK1和Parkin蛋白表达显著下调,且与线粒体通透性转换孔(mitochondrial permeability transition pore,mPTP)开放及其相关线粒体凋亡途径激活有关[57]。哺乳动物不育系20样激酶1是新近发现的线粒体自噬上游调节因子,该基因消融可上调Parkin蛋白表达,恢复Parkin介导的线粒体自噬,从而减轻高脂膳食诱导的肝损伤以维持肝细胞存活[55]。与之一致的是,给予槲皮素处理亦可通过增强PINK1/Parkin依赖性线粒体自噬以减轻10周高脂饮食诱导的小鼠肝功能紊乱[56]。此外,对ob/ob小鼠施加4周二甲双胍干预可通过上调Parkin介导的线粒体自噬而抑制p53介导的肝细胞凋亡[60]。然而,Kim等[61]报道指出,与野生型小鼠相比,同期给予6周高脂饮食喂养的Parkin基因敲除小鼠呈现出肝脂肪变性、肥胖和胰岛素抵抗程度明显减轻。究其机理,Parkin是通过泛素介导的脂肪酸转位酶CD36在肝内稳定表达而发挥上述作用。此外,在为期一周的高脂饮食喂养模型中,Parkin基因敲除小鼠的肠道脂质吸收受损,表现为粪便脂质增加和血浆甘油三酯减少,胰岛素敏感性增强[62]。上述研究结果突出了Parkin除介导线粒体自噬外,在调节脂质吸收方面亦发挥重要作用,Parkin缺失可能通过减少肠道脂质吸收而减轻高脂膳食诱导的肝损伤。
2.2.2 Bnip3与NAFLD
首先,Bnip3介导的线粒体自噬在调节肝脏脂质代谢方面具有重要作用[63,64]。Glick等[63]报道指出,常规膳食喂养的Bnip3基因敲除小鼠肝脏脂质合成明显增加,呈现出肝脏脂肪变性,并伴随AMPK活性下降、ATP水平和成脂酶表达升高,肝线粒体表现出膜电位降低、结构异常和耗氧量减少等功能紊乱;此外,他们给予禁食处理后观察到野生型小鼠肝脏Bnip3和LC3-Ⅱ蛋白表达显著升高,而Bnip3基因敲除小鼠肝脂肪酸β-氧化明显减弱,脂质囊泡数量增多且体积变大,同时线粒体膜蛋白细胞色素C氧化酶亚基IV(cytochrome c ox-idease subunitⅣ,COXIV)表达显著升高,以上结果提示能量限制(禁食)在一定程度上能激活Bnip3依赖性线粒体自噬,且Bnip3可通过介导受损线粒体的清除以维持肝脏线粒体正常生理功能,从而有效抑制肝脂质蓄积变性。此外,Gong等[64]在BRL大鼠肝细胞内亦证实木通皂甙D能明显上调Bnip3和p-AMPK蛋白表达,并诱导LC3-Ⅱ线粒体定位增加,而施加AMPK抑制剂Compound C则可逆转上述结果,最终导致NAFLD相关肝脂肪变性。以上研究结果表明在肝细胞内能量开关AMPK的活化对于Bnip3介导线粒体自噬必不可少,并且靶向激活Bnip3可能是防治肝脂肪变性的有效策略。
其次,Bnip3还被认为是调节线粒体自噬和NAFLD进展之间平衡的关键[58,65,66]。Rosa-Caldwell等[65]报道,饲喂高脂膳食小鼠肝Bnip3 mRNA和蛋白表达以及LC3-Ⅱ蛋白表达均低于常规膳食小鼠。Li等[66]观察到,线粒体定位的SIRT3过表达可通过激活细胞外信号调节激酶(extracellular signal-regulate kinase,ERK)/cAMP反应元件结合蛋白(cAMP response-ele-ment binding protein,CREB)信号通路上调Bnip3蛋白表达及其介导的线粒体自噬,从而抑制棕榈酸诱导的肝细胞凋亡。此外,Zhou等[58]研究发现,NAFLD状态下孤核受体亚家族4A型成员1(orphan nuclear recep-tor subfamily 4 group A member 1,NR4A1)表达增加并激活DNA依赖性蛋白激酶催化亚单位(DNA-de-pendent protein kinase catalytic subunit,DNA-PKcs)/p53信号通路,一方面促进发动蛋白相关蛋白1(dyna-min-related protein 1,Drp1)线粒体定位,致使线粒体过度分裂,另一方面明显下调Bnip3的mRNA和蛋白表达,最终引起线粒体功能障碍(线粒体膜电位降低、mPTP开放、氧化应激、Ca2+超载、线粒体呼吸衰竭和ATP缺乏)及自噬受阻而加重NAFLD。相反,给予褪黑素处理能通过抑制NR4A1/DNA-PKcs/p53信号途径而减弱Drp1介导的线粒体分裂,同时明显促进Bnip3依赖性线粒体自噬,改善线粒体功能障碍,进而延缓NAFLD的发生发展。综上所述,Bnip3通过介导线粒体自噬过程而参与调节NAFLD相关的肝脂肪变性,且受AMPK、SIRT3/ERK/CREB、NR4A1/DNA-PKcs/p53等多条信号通路调控。在NAFLD进程中,不同的信号分子通过在转录和翻译水平调控Bnip3的表达而诱导肝线粒体功能紊乱和分裂/自噬功能失衡。目前,在任何特定的病理生理条件下,Bnip3介导的线粒体自噬和肝脂肪变性之间的主要信号通路仍然是未知的。基于此,Bnip3在介导NAFLD相关线粒体自噬中的调控机制有待进一步研究阐明。
3 运动通过改善自噬功能障碍防治NAFLD
如前所述,NAFLD的发生发展与自噬功能障碍相关,改善自噬活性现已被公认为NAFLD的一种潜在防治策略[13]。目前,防治NAFLD最有效的非药理手段仍主要是生活方式干预(通常包括运动、热量限制和减重)[1],其中运动和热量限制是两种主要的自噬诱导方式[13],这很可能是二者改善肝功能不全和脂肪变性的作用机制。大量研究结果表明,长期规律运动可通过激活肝脏能量开关因子AMPK促进细胞自噬[8,67,68]和线粒体自噬[69],改善肝脂肪变性与线粒体功能障碍,从而有效防治NAFLD。
3.1 运动诱导细胞自噬以防治NAFLD
目前研究表明,规律运动可通过激活细胞自噬而预防NAFLD的发生发展[6,8,9](表1)。Wang等[9]对小鼠采取24周高脂饮食喂养联合跑台运动干预,结果显示,与高脂安静组相比,高脂运动组小鼠肝脏LC3-Ⅱ/LC3-Ⅰ的比值虽无明显改变但p62蛋白表达显著下降,肝脂肪变性和纤维化程度明显减轻,提示24周持续中等强度有氧运动可能通过上调肝细胞自噬通量而有效预防高脂饮食诱导的NAFLD。再如,Pi等[6]对小鼠施加高脂膳食饲喂联合游泳训练,观察到小鼠经12周高脂饮食喂养后体重明显增加并伴血脂异常(甘油三酯、总胆固醇显著升高),且肝内脂滴大量形成,同期运动干预可明显下调肝脂肪变性和p62蛋白表达并上调LC3蛋白表达,表明12周游泳训练通过激活肝细胞自噬而促进脂质分解并抑制其变性。与Pi等[6]研究相似,Li等[8]采取高脂饮食喂养诱导NAFLD建模的同时联合游泳运动干预,结果显示16周游泳运动可通过上调AMPK/SIRT1通路增强小鼠肝细胞自噬和脂肪酸氧化能力,进而有效预防高脂膳食诱导的NAFLD。此外,适当的溶酶体降解能力是维持自噬通量和降解脂滴所必需的[70],Pi等[6]的研究还显示12周游泳训练能逆转同期高脂膳食所致的肝溶酶体功能受损,显著上调与溶酶体功能(维持溶酶体酸化、增加溶酶体蛋白水解)相关的V-ATP酶亚基ATP6V0D1以及溶酶体组织蛋白酶D(cathepsin D,CTSD)的蛋白表达;Li等[8]的研究亦观察到16周游泳运动可明显上调小鼠肝溶酶体相关膜蛋 白1(lysosomal-associated membrane protein 1,LAMP1)蛋白表达,以上结果提示长期规律运动亦可通过改善溶酶体功能促进自噬降解,从而预防高脂饮食诱导的NAFLD。
另外,大量临床前研究也已证实规律运动对NAFLD模型下细胞自噬的重要作用[67,68,71](表1)。Tang等[71]通过10周高脂饮食诱导NAFLD小鼠肝脏LC3-Ⅱ/LC3-Ⅰ的比值明显下调且p62蛋白表达显著上调,引起肝脂肪变性。然而10周游泳训练通过上调LC3-Ⅱ/LC3-Ⅰ的比值,并下调p62蛋白表达,从而减轻肝脏脂肪变性程度,促进NAFLD的良性转归。Gao等[67]对高脂膳食构建的NAFLD模型大鼠进行8周运动干预,结果显示,持续中等强度跑台运动能明显上调肝AMPK、ULK1磷酸化表达和LC3-Ⅱ/LC3-Ⅰ的比值,并显著下调p62蛋白表达和肝脂肪变性程度,提示持续中等强度有氧运动能通过激活AMPK/ULK1途径而改善NAFLD肝损伤和自噬功能障碍。同样,Ghareghani等[68]采取13周高脂膳食饲喂诱导NAFLD小鼠建模,随后施加10周跑台运动干预,除观察到运动后小鼠肝脂肪变性、肝LC3-Ⅱ/LC3-Ⅰ的比值和p62蛋白表达明显改善以外,还检测到自噬相关因子ATG5、ATG7蛋白表达和Beclin-1 mRNA表达以及AMPK磷酸化显著升高,而mTOR磷酸化表达明显下降,以上结果表明规律性有氧运动能有效改善高脂膳食所致的NAFLD肝脂质异常蓄积,其机理可能与活化的AMPK介导其下游mTOR和/或ULK1磷酸化进而增强ATG7相关的细胞自噬有关。因此,靶向上调AMPK等关键能量开关分子表达的策略或许将有助于防治NAFLD。与规律运动介导的预防效应相似的是,Gao等[67]和Ghareghani等[68]的研究亦分别检测到LAMP1、LAMP2蛋白表达和LAMP2 mRNA表达明显上调,提示规律运动可通过改善溶酶体功能以促进细胞自噬或为NAFLD治疗的重要途径。

表1 运动对NAFLD肝细胞自噬及线粒体自噬的影响

(续表1)
3.2 运动诱导线粒体自噬以防治NAFLD
除细胞自噬外,运动还能通过改善线粒体自噬而防治NAFLD(表1)。赵军等[7]采取高脂喂养诱导NAFLD建模的同时联合23周中等强度跑台运动干预,结果显示,大鼠经高脂膳食饲喂16周成功建立NAFLD模型,引起脂肪型肝炎和肝纤维化的发生,而在高脂喂养之前7周及高脂喂养期间施加有氧运动干预可明显上调肝脏线粒体增殖和自噬调控因子SIRT1、过氧化物酶体增殖物激活受体γ共激活因子1α(peroxisomeprolif-erator-activated receptorγcoactivator-1α,PGC-1α)、COXⅣ和Parkin的mRNA表达以及LC3-Ⅱ/LC3-Ⅰ的比值,且显著下调肝脂质蓄积和丙二醛含量。上述研究结果与Pi等[6]、Li等[8]的研究结果一致,表明长期规律运动可有效预防高脂饮食经氧化应激途径所诱导的NAFLD,其机理可能与激活肝细胞内SIRT轴而增强线粒体增殖和自噬有关。
此外,不少研究亦证实规律运动可通过PINK1/Parkin依赖性线粒体自噬促进NAFLD的良性转归[57,69]。例如,Gonçalves等[57]通过9周高脂饮食构建NAFLD大鼠模型,随后进行8周跑台运动干预可显著上调肝脏PINK1和Parkin蛋白表达,LC3-Ⅱ亦呈现出升高趋势,且明显下调肝脂肪变性,提示中等强度跑台运动能通过增强线粒体自噬有效改善NAFLD肝损伤。再如,Guarino等[69]报道指出,12周胆碱缺乏高脂饮食诱导的NAFLD模型小鼠在完成8周跑台运动后呈现出肝脏脂肪变性明显下调,且能量开关AMPK和线粒体自噬蛋白PINK1磷酸化表达以及LC3-Ⅱ/LC3-Ⅰ的比值显著上调。上述研究结果表明,长期规律运动可通过激活PINK1/Parkin介导的线粒体自噬而延缓NAFLD发展进程,且AMPK在调控线粒体自噬改善肝脏脂质异常蓄积等方面可能具有重要作用。
综上所述,长期运动不仅可通过活化的AMPK介导其下游mTOR和/或ULK1磷酸化进而增强ATG7相关的细胞自噬,还能通过激活AMPK和/或SIRT1从而上调PINK1/Parkin依赖性线粒体自噬,促进肝脏脂质分解,逆转脂质堆积所致的自噬流阻断,并改善高脂膳食等诱导的肝脂肪变性,从而有效预防和/或延缓NAFLD至NASH等进行性病症的发生发展。上述健康效益对于NAFLD患者运动处方的个性化设计具有重要的理论指导意义(图1)。未来可开展以防治NAFLD为目的的大规模随机对照试验,针对不同年龄、不同病程的NAFLD患者制订合理的运动处方,以进一步厘清运动通过诱导细胞(线粒体)自噬改善NAFLD的具体分子机制。
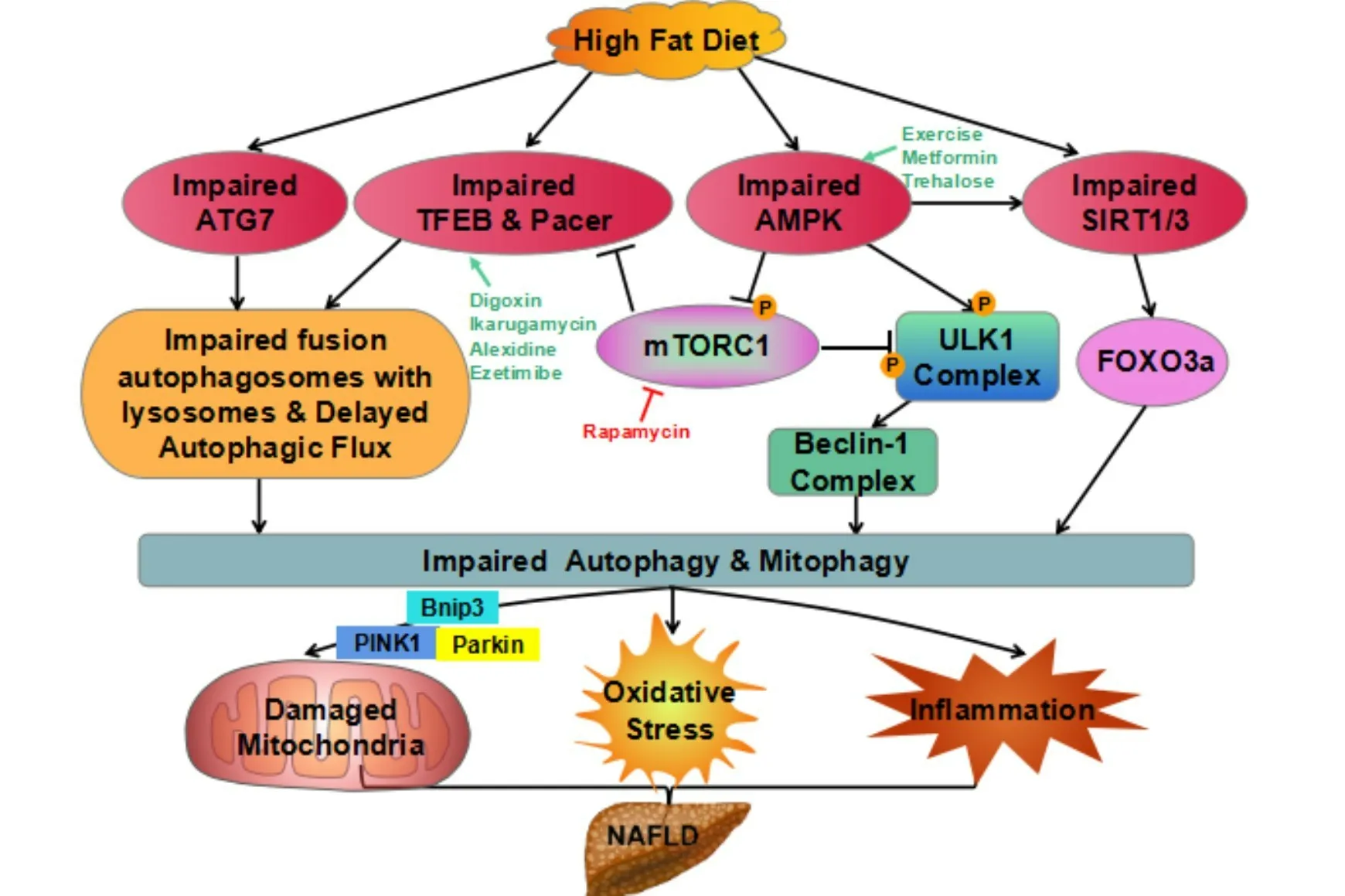
图1 NAFLD的潜在诱发机制与干预靶点
4 总结
鉴于自噬可通过介导肝细胞内脂解及受损线粒体清除以抑制肝内脂质蓄积变性,故自噬功能障碍被视为NAFLD及其相关脂代谢异常的发病机制。其中,AMPK-mTORC1-ULK1、ATG7和Ca2+等信号途径以及PINK1/Parkin和Bnip3等线粒体自噬蛋白分别参与调节NAFLD条件下受损的细胞自噬及线粒体自噬。因此,通过运动、AMPK激活剂(如二甲双胍和海藻糖)、mTORC1抑制剂雷帕霉素、TFEB激活剂(如地高辛、斑鸠霉素、阿来西定、依折麦布)和热量限制等方式激活自噬以维持肝脏脂质代谢稳态可能是防治NAFLD的关键因素。规律运动不仅可通过活化的AMPK介导其下游mTOR和/或ULK1磷酸化进而增强ATG7相关的细胞自噬,还能通过激活AMPK和/或SIRT1而上调PINK1/Parkin依赖性线粒体自噬,逆转脂质沉积所致的自噬流阻断,从而有效预防和/或延缓NAFLD的发生发展。目前,有如下关键问题尚待阐明:(1)NAFLD的复杂病因和病理生理学机制(包括遗传和环境因素影响)致使难以基于离体细胞和在体动物实验结果对人体研究进行推断,目前有关运动调节自噬途径防治NAFLD的研究成果集中于动物实验,尚缺乏人群方面的研究;(2)自噬相关基因ATG7在肝脂肪变性中的作用仍存在争议,需进一步明确其促进/抑制作用及其与Ca2+信号的关系;(3)Bnip3介导线粒体自噬在运动防治NAFLD进程中的作用尚不清楚,需探明除PINK1/Parkin外是否还有更多的线粒体自噬相关蛋白参与其中;(4)AMPK在细胞自噬及线粒体自噬中的作用还有待深入研究,可构建AMPK敲除/敲低/过表达的特异性基因模型探究运动、药物和热量限制等防治手段调节代谢的分子机制及评估AMPK依赖性和非依赖性自噬调控效应。厘清上述问题将为进一步优化NAFLD及其相关疾病的临床诊治方案提供理论与实验基础。
