固相萃取-离子色谱法测定坎地沙坦酯原料药中叠氮化钠和四丁基溴化铵的残留量
2022-06-24苗华明刘丽霞徐晓蕾
李 晶,刘 敏,苗华明,刘丽霞,徐晓蕾
(迪沙药业集团国家认定企业技术中心,威海 264200)
坎地沙坦酯是一种新型降压药,属于选择性血管紧张素Ⅱ受体(ATI)拮抗剂。叠氮化钠和四丁基溴化铵作为坎地沙坦酯传统工艺路线中的关键催化剂,在坎地沙坦酯的合成过程中发挥着重要作用。叠氮化钠是剧毒物质,可抑制细胞色素氧化酶和其他酶,造成电子传递过程中断,从而损害神经系统[1]。四丁基溴化铵对皮肤、眼睛和呼吸系统有刺激性作用,且具有基因毒性警示结构。为了保障药物的质量安全,中华人民共和国药典(2015年版)和相关指导原则[2]规定,要严格控制两者在药物中的残留量。由于未查到这两种杂质的具体毒性数据,参照国际人用药品注册技术协调会(ICH)M7指导原则[3-4],两种杂质的每日最大服用量不得超过1.5μg,而坎地沙坦酯的日最大服用量为32 mg,可计算得到四丁基溴化铵和叠氮化钠的质量分数不得超过0.004 7%。而叠氮化钠和四丁基溴化铵在药物中残留量较低,对检测灵敏度要求高,因此开发一种高灵敏度的分析方法至关重要。
目前,测定叠氮化钠和四丁基溴化铵的方法有液相色谱-质谱法[5-7]、离子色谱法[8]。其中液相色谱-质谱法操作复杂,对仪器要求高;四丁基溴化铵、叠氮化钠均为离子型化合物,水解后会生成Br-、N 3-,经阴离子淋洗系统(氢氧化钾体系)淋洗后,可用附电导检测器的离子色谱法检测[9-10]。
鉴于此,本工作用水提取样品中的两种目标化合物,用HyperSep C18固相萃取小柱消除坎地沙坦酯的影响,采用附电导检测器的离子色谱法进行抑制电导检测。该方法操作简便、灵敏度高、回收率好,可用于坎地沙坦酯原料药中叠氮化钠和四丁基溴化铵残留量的测定。
1 试验部分
1.1 仪器与试剂
ICS-5000型离子色谱仪,配电导检测器;DL-1000D 型智能超声仪;HC2062 型高速离心机;MS105DU 型电子天平;Milli-Q 型超纯水仪;HyperSep C18固相萃取小柱(200 mg/3 mL)。
叠氮化钠对照品储备溶液:7.25 mg·L-1,取叠氮化钠对照品14.5 mg,用水溶解并定容至100 mL,分取5 mL置于100 mL容量瓶中,用水稀释至刻度,摇匀。
Br-对照品 储备溶 液:1.00 mg·L-1,取1 000 mg·L-1Br-对照品溶液1 mL置于100 mL容量瓶中,用水稀释至刻度。分取1 mL 置于10 mL容量瓶中,加水稀释至刻度,摇匀。
NO3-对照品 储备溶 液:1.00 mg·L-1,取1 000 mg·L-1NO3-对照品溶液1 mL 置 于100 mL容量瓶中,加水稀释至刻度。分取1 mL置于10 mL容量瓶中,加水稀释至刻度,摇匀。
系统适用性溶液:分别取1 mL 叠氮化钠对照品储备溶液、1.2 mL Br-对照品储备溶液和4.7 mL NO3-对照品储备溶液,置于同一10 mL容量瓶中,加水稀释至刻度,摇匀,即得含0.725 mg·L-1叠氮化钠、0.12 mg·L-1Br-和0.47 mg·L-1NO3-的系统适用性溶液。
叠氮化钠对照品纯度为99.6%;1 000 mg·L-1Br-对照品溶液;1 000 mg·L-1NO3-对照品溶液;四丁基溴化铵为分析纯;试验用水为超纯水(电阻率为18.2 MΩ·cm)。
坎地沙坦酯原料药样品由本公司自制。
1.2 仪器工作条件
IonPac AS18阴离子交换色谱柱(250 mm×4 mm,7.5μm);柱温35℃;检测方式为抑制电导检测,抑制电流99 m A;电导池温度35 ℃;进样体积200μL;淋洗液为氢氧化钾溶液,流量1.0 mL·min-1。梯度淋洗程序:0~25 min时,氢氧化钾溶液浓度为9 mmol·L-1;25~38 min时,氢氧化钾溶液浓度由9 mmol·L-1升至40 mmol·L-1;38~45 min 时,氢氧化钾溶液浓度由40 mmol·L-1降至9 mmol·L-1。
1.3 试验方法
取坎地沙坦酯原料药样品约100 mg,加入10 mL水,超声15 min(每隔5 min强力振摇30 s)。分取约5 mL 置于10 mL 离心管中,以转速10 000 r·min-1离心10 min。上清液过0.22μm滤膜,滤液过HyperSep C18固相萃取小柱(使用前先用3 mL甲醇活化,再用6 mL水冲洗干净),收集洗脱液,经0.22μm 滤膜过滤,滤液即为供试品溶液,按照仪器工作条件测定。
2 结果与讨论
2.1 固相萃取步骤的影响
在样品检测时,发现供试品溶液中含量较高的坎地沙坦酯会吸附在色谱柱上且很难被洗脱掉,这将导致色谱柱柱压变化,柱效降低。为了延长色谱柱使用寿命,试验选择在进样前增加固相萃取步骤,以降低坎地沙坦酯对色谱柱的损害。
2.2 色谱条件的选择
2.2.1 检测器
离子色谱常用的检测器有电导检测器和电化学检测器,其中电导检测器主要适用于离子型化合物,而四丁基溴化铵和叠氮化钠均为离子型化合物,因此试验选择附电导检测器的离子色谱进行抑制电导检测。在此检测条件下,背景电导以及基线噪声较低,检测灵敏度大大提高[11-14]。
2.2.2 色谱柱和柱温
试验考察了规格相同的不同阴离子色谱柱Ion-Pac AS11和Ion Pac AS18对Br-和N3-分离效果的影响。结果显示:IonPac AS11 柱不能实现Br-和N3-的基线分离;而采用IonPac AS18 柱时,色谱峰峰形对称,分离度良好。因此,试验选择的阴离子色谱柱为IonPac AS18柱。
试验进一步考察了柱温分别为30,35 ℃时对Br-和N3-分离效果的影响。结果显示,两种温度下Br-和N3-均能完全分开,考虑到35℃时仪器压力较小,试验选择的柱温为35 ℃。
2.2.3 淋洗液浓度和淋洗方式
试验考察了淋洗液氢氧化钾溶液初始浓度分别为5,9 mmol·L-1时对Br-和N3-分离效果的影响。结果显示,当淋洗液初始浓度为9 mmol·L-1时,N3-前后杂质峰不干扰其出峰,因此试验选择淋洗液氢氧化钾溶液的初始浓度为9 mmol·L-1。
水中常见的阴离子有NO2-、SO42-、HCO3-、NO3-、HCOO-,其中NO3-色谱峰位置和Br-、N3-的距离较近,可能影响Br-和N3-检测结果的准确度。为保证Br-、N3-和NO3-能完全分离,试验选择梯度淋洗程序,即先采用较低浓度淋洗液淋洗25 min,保证Br-、N3-和NO3-完全出峰,然后用较高浓度淋洗液冲洗13 min,保证样品带进去的离子都被洗脱出来,最后平衡7 min,详见1.2节。
2.3 四丁基溴化铵离子化程度的影响
四丁基溴化铵是一种有机盐,其离子化程度会影响检测结果的准确度。为了考察四丁基溴化铵离子化程度对测定结果的影响,分别配制0.82 mg·L-1四丁基 溴化铵溶液(Br-的质量 浓度约0.20 mg·L-1)和0.20 mg·L-1Br-对照品溶液,按照仪器工作条件测定。结果显示,两种溶液中Br-的保留时间分别为19.843,19.797 min,峰面积分别为0.147,0.155μS·min,两者保留时间和峰面积基本一致,以线性回归方程计算得四丁基溴化铵溶液中Br-的质量浓度为0.19 mg·L-1,所得Br-的回收率约为95.0%,说明四丁基溴化铵的离子化程度比较完全,检测结果准确度较高。
2.4 系统适用性试验
按照仪器工作条件测定空白溶剂、供试品溶液和系统适用性溶液,色谱图见图1。
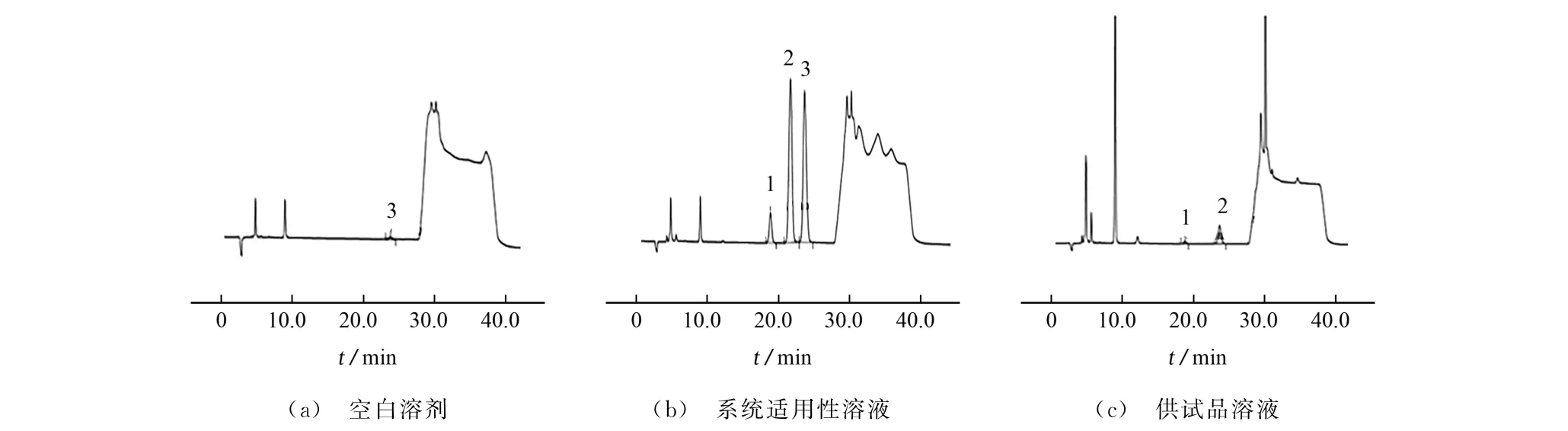
图1 空白溶剂、系统适用性溶液和供试品溶液的色谱图Fig.1 Chromatograms of blank solvent,system suitability solution and test solution
结果显示:Br-、N3-和NO3-均可实现基线分离,NO3-与N3-分离度为2.42;空白溶剂对Br-、N3-的检测无干扰,供试品溶液中其他杂质对Br-、N3-的检测无干扰,说明该方法专属性较好。
对系统适用性溶液连续进样6 次,计算杂质NO3-保留时间和峰面积的相对标准偏差(RSD),所得保留时间的RSD 不大于0.010%,小于1.0%,峰面积RSD 不大于0.5%,小于2.0%,满足规定的检测要求。
2.5 标准曲线、检出限和测定下限
以3倍信噪比(S/N)计算检出限(3S/N),N3-和Br-的检出限分别为0.00070,0.0014 mg·L-1。以10倍信噪比计算测定下限(10S/N),N3-和Br-的测定下限分别为0.00233,0.0048 mg·L-1。
用水逐级稀释Br-对照品储备溶液和叠氮化钠对照品储备溶液,以测定下限作为第一个浓度点,分别以0.466mg·L-1和0.120mg·L-1作为N3-和Br-的100%杂质限度,再分别配制25%,50%,100%,150%,200% 杂质限度的浓度点,即得0.002 33,0.116,0.233,0.466,0.699,0.932 mg·L-1的N3-对照品 溶液系列和0.0048,0.030,0.060,0.120,0.180,0.240 mg·L-1的Br-对照品溶液系列。按照仪器工作条件测定,以N3-和Br-的质量浓度为横坐标,其对应的峰面积为纵坐标绘制标准曲线。结果显示:N3-和Br-的标准曲线的线性范围分别为0.002 33~0.932 mg·L-1和0.004 8~0.240 mg·L-1,线性回 归方程 分别为y=1.336x+6.600×10-3和y=8.006×10-1x-2.500×10-3,相关系数均为0.999 8。
2.6 精密度和回收试验
按照试验方法对空白样品进行低、中、高等3个浓度水平的加标回收试验,各浓度水平分别平行测定3,6,3次,计算回收率和中浓度水平下测定值的RSD,结果见表1。
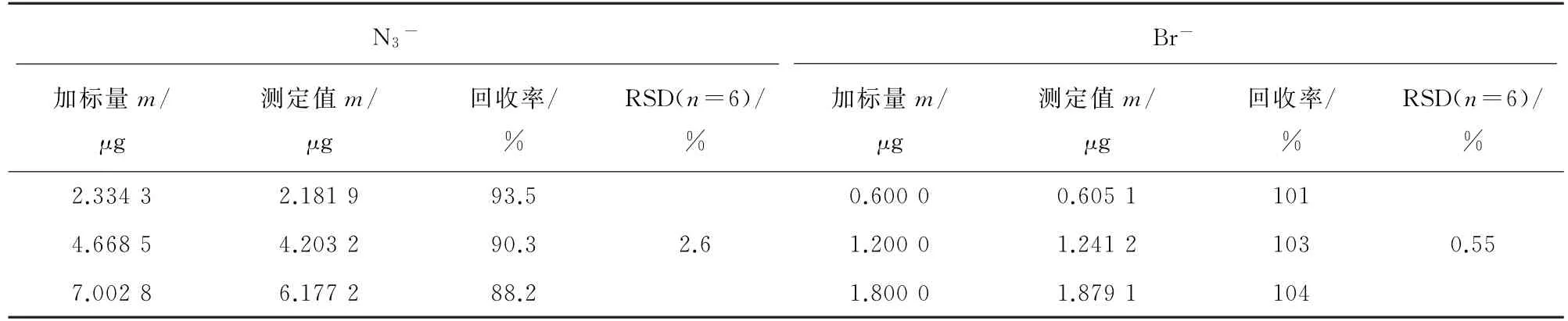
表1 精密度和回收试验结果Tab.1 Results of tests for precision and recovery
由表1可知,N3-和Br-的回收率分别为88.2%~93.5%和101%~104%,测定值的RSD 分别为2.6%和0.55%,说明方法的准确度和精密度均较好。
2.7 稳定性试验
将加标供试品溶液(N3-和Br-的加标量分别为4.668 5,1.200 0μg)分别在室温下放置0,2,4,6,8 h,按照仪器工作条件测定,计算峰面积的RSD。结果显示,N3-和Br-峰面积的RSD 分别为2.3%和1.1%,说明供试品溶液在8 h内稳定。
2.8 耐用性试验
略微调整淋洗液浓度、柱温、流量参数,比较了在不同色谱条件下系统适用性溶液中Br-、N3-和NO3-分离度的变化,结果见表2。
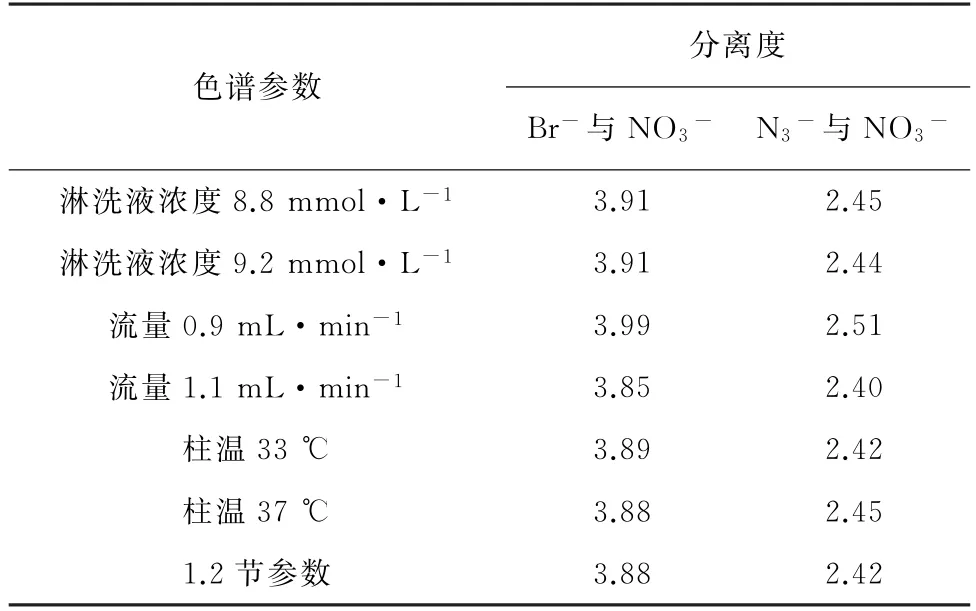
表2 耐用性试验结果Tab.2 Results of test for durability
由表2 可知,略微调整色谱参数后,Br-与NO3-、N3-与NO3-之间的分离度均大于1.50,说明方法耐用性较好。
2.9 样品分析
按照试验方法分析坎地沙坦酯原料药小试、中试以及批量生产产品各3批,分析结果见表3。
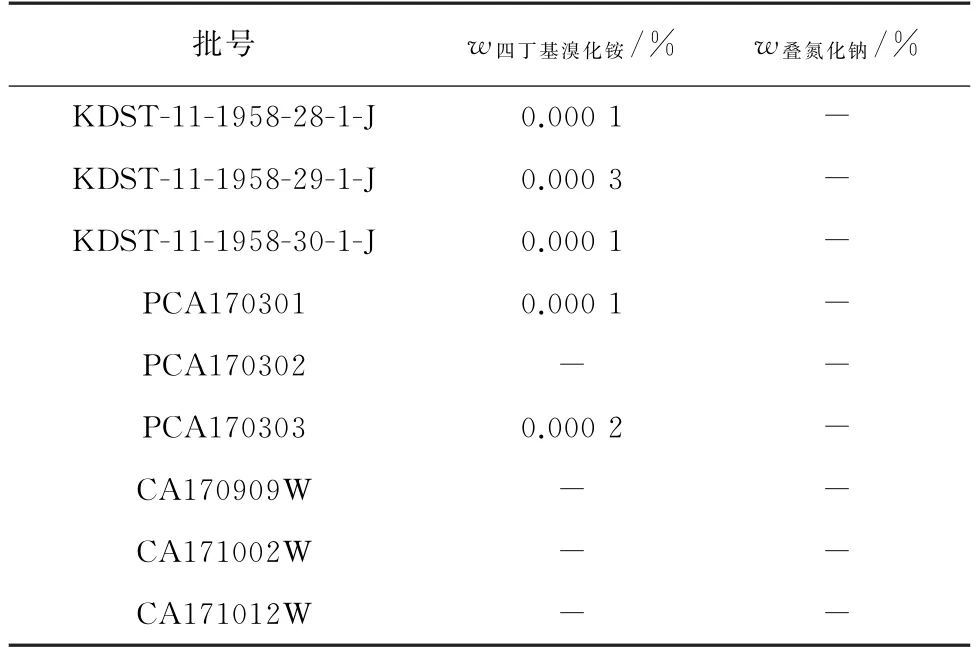
表3 样品分析结果Tab.3 Analytical results of the samples
结果表明,开发的检测方法可以满足生产检测的需求,同时样品中的其他杂质亦不干扰Br-、N3-的测定。
本工作采用离子色谱法同时测定坎地沙坦酯原料药中叠氮化钠和四丁基溴化铵的含量,原料药以及溶剂水中其他杂质离子对Br-、N3-干扰小。方法专属性和灵敏度较好,准确度、稳定性、耐用性均较好,可用于坎地沙坦酯原料药质量的监控。
