Primary myelofibrosis with thrombophilia as first symptom combined with thalassemia and Gilbert syndrome:A case report
2022-06-23GuzailinuerWufuerKaisaerWufuerTuBaTaoCuiLingTaoLingFuMingMaoMingHuiDuan
lNTRODUCTlON
Thrombophilia refers to heredity or acquired defects in blood clotting,such as defects existing in anticoagulant proteins,blood coagulation factors,and fibrinolytic proteins,or acquired risk factors resulting in a higher tendency for thromboembolism.Hereditary antithrombin(AT)deficiency is one of the major risk factors for hereditary thrombophilia.This hereditary deficiency is caused by a mutation in the AT coding gene SERPINC1.Not all people with AT gene mutations will experience hereditary AT deficiency.The occurrence of hereditary AT deficiency is also affected by acquired risk factors.Under the dual effect of a heredity gene mutation and acquired risk factors,the incidence of hereditary AT deficiency will be increased distinctly.We hereby report the case of a patient with thrombophilia with a hereditary AT deficiency but no individual or family thrombosis history.Genetic testing revealed a heterozygous SERPINC1 mutation and a JAK2V617-positive myeloproliferative neoplasm(MPN)as a potential acquired risk factor for venous thrombosis.At the same time,the family was confirmed to have UGT1A1 and β-thalassemia gene mutations.
CASE PRESENTATlON
Chief complaints
A 46-year-old Han man had thrombophilia,an obviously enlarged spleen,and a history of jaundice for many years.
8. Alone in the wood: Julius Heusher states that the woods represent the loss of security and previous values (Heuscher 1974).Return to place in story.
History of present illness
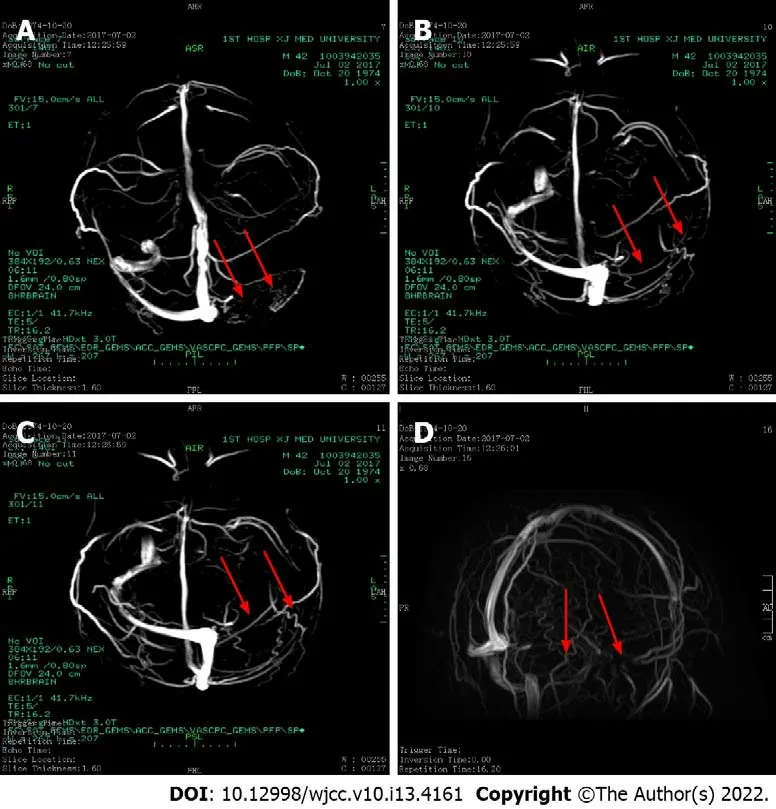
The patient was admitted to the hospital on August 4,2019 due to “scleral yellow staining for more than 30 years and recurrent venous thrombosis for 2 years”.Since the age of 10 years,the patient had noted yellow sclera and dark yellow urine,but he denied abdominal pain,abdominal distension,nausea,and vomiting.Because the symptoms did not disturb his daily life or studying,he did not paid attention to them.In 2005,he underwent cholecystectomy because of a "gallstone".In June 2017,he experienced abdominal distension,headache,and diarrhea with associated discomfort.After a head computed tomography(CT)and magnetic resonance(MR)examination,the diagnosis was "the left sigmoid sinus and transverse sinus have abnormal signals and abnormal enhancement,suggesting venous sinus thrombosis”(Figure 1).
History of past illness
No history of personal or family thrombosis.
Personal and family history
No history of personal or family thrombosis.
Physical examination
The physical examination revealed that the patient did not have an anemia appearance,but he had slight yellow sclera and splenomegaly,without superficial lymph node enlargement.
Laboratory examinations
In China,Li
[21]reported the case of a patient who was positive for the JAK2/V617F mutation and a UGT1A1 mutation and had MPN combined with Gilbert syndrome,an increased WBC count and PLT count,mild anemia,and splenomegaly.Bone marrow and pathological biopsy suggested hyperactive hyperplasia and increased megakaryocytes.Blood biochemical tests showed severe jaundice and mainly increased indirect bilirubin.UGT1A1 gene sequencing revealed an insertion mutation in exon 1,from(TA)6TAA to(TA)7TAA,that is,from wild-type UGT1A1*1 to UGT1A1*28,and a missense mutation,heterozygous C.211G>A,UGT1A1*6,resulting in a decrease in glucuronide transferase activity.The patient and his son both had polymorphic missense mutations in the promoter and noncoding regions.The patient's son did not develop the disease,and the patient was the only case reported in China.Engelborghs
[22]reported the case of a 15-year-old boy with mild thalassemia and thrombosis.The patient was heterozygous for thalassemia,and he had recurrent transient ischemic attacks.Because the patient experienced resistance to activated protein C,his condition was suspected to be due to factor V heterozygosity and a slight decrease in proteins C and S in plasma levels.
The Prince turned sharply round and to his horror saw a huge Giant approaching with mighty strides, crying fiercely-- Who has made my lion whistle I should like to know? I have, replied Prince Vivien boldly, but I can answer for it that he will not do it again! At these words the Giant began to howl and lament94
Gilbert syndrome,also known as hereditary unbound bilirubinemia,is usually clinically characterized by mild to moderate increases in simple unbound bilirubin,which is caused by uridine diphosphate glucuronide transferase gene mutations[16].In addition to intermittent jaundice attacks,most patients with Gilbert syndrome are asymptomatic and have normal physical examination results.Gilbert syndrome usually begins in adolescence,when changes in the concentration of sex steroid hormones affect bilirubin metabolism,leading to an increase in plasma bilirubin concentration.As a result,the disease is rarely diagnosed before puberty.Gilbert syndrome is more common in men,possibly because men's daily bilirubin production levels are higher than those of females[17].Some patients have nonspecific symptoms,such as general malaise,abdominal discomfort,or fatigue,and the relationship between these complaints and rising plasma bilirubin concentration is unclear.A case report showed that some patients with Gilbert syndrome had severe unconjugated hyperbilirubinemia.Such patients usually also have other mutations in the UGT1A1 promoter region,are heterozygous carriers of Crigler-Najjar type structural mutations,or have coexisting diseases that are prone to hyperbilirubinemia(for example,diseases leading to hemolysis).A report of two such patients(one patient with hereditary spherocytosis and the other with other mutations in UGT1A1)showed that hyperbilirubinemia was successfully treated with rifampin,the function of which is the induction of UGT1A1[18].Lee
[19]reported a 28-year-old male patient with gallstones and splenomegaly,combined with hereditary spherocytosis and Gilbert syndrome.Jiang
[20]reported a case in which a patient had dual hereditary jaundice with Dubin-Johnson syndrome and Gilbert syndrome mutations at the same time in China,and this has been the only case reported in China.
Wufuer G,Wufuer K,Ba T,Cui T,and Tao L contributed equally to this work;Fu L and Mao M designed the research study;Duan MH analyzed the data and wrote the manuscript;all authors have read and approved the final manuscript.
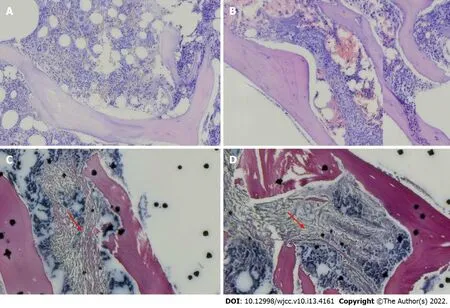
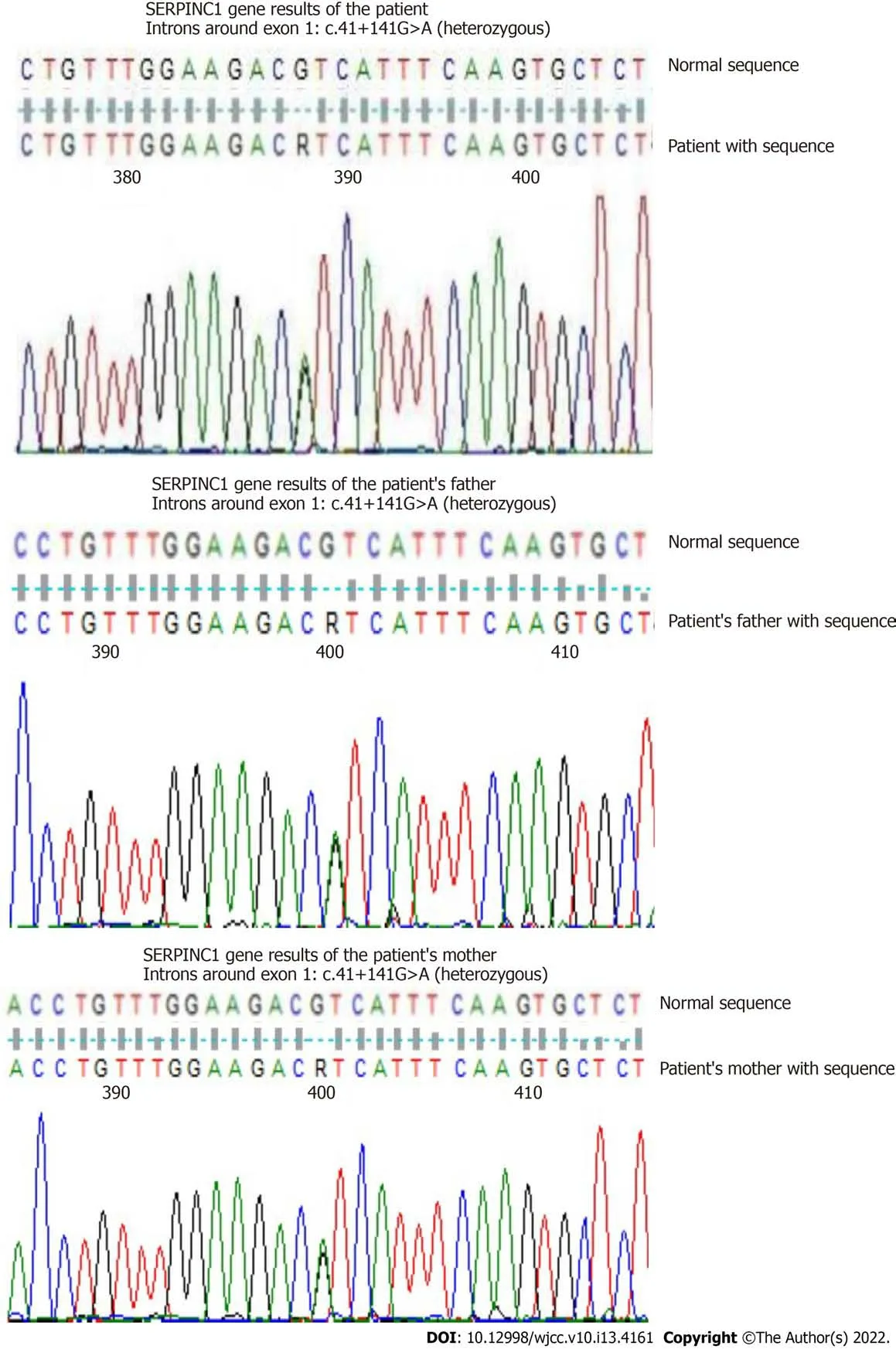
The portal vein was 12 mm in diameter,and splenomegaly was observed(68 mm × 205 mm).After cholecystectomy,the common bile duct was not dilated.The pancreas and kidneys had no spaceoccupying lesions.Ultrasound of the portal vein showed portal vein thrombosis with collateral angiogenesis.Ultrasound of the arteries and veins in the upper and lower limbs revealed no abnormalities.Hemoglobin electrophoresis revealed 1.8% of fetal HB(HbF),5.7% of glycated HB(HbA2),and 55%(> 75%)of methemoglobin.In addition,the following test results were observed:Erythrocyte osmotic fragility test(starting hemolysis),4.4 g/L(3.8-4.6);complete hemolysis test,3.2 g/L(2.8-3.2);and G-6PD,2628.11.In a right posterior superior iliac spine bone marrow smear,the bone marrow had active nuclear cell proliferation,and the ratio of granulocytes to erythrocytes was 1.5:1.Erythrocyte hyperplasia was active,accounting for 44%.Middle and late erythrocyte hyperplasia was dominant,there was no abnormal morphology,and the sizes of some mature erythrocytes were different.The number of megakaryocytes was normal and functional.Marrow immune typing was not special.In bone marrow biopsy,the ratio of hematopoietic tissue to adipose tissue in bone marrow was approximately 5:1,and the ratio of granulocytes to erythrocytes was approximately 1:2.Erythrocytes showed active hyperplasia,and the features of granulocytes and erythrocytes were not abnormal.The trabeculae showed looser and crossed reticular fibers,4-8 megakaryocytes/high-power field,and a fibrosis grade of MF-1(Figure 2).The patient had a normal chromosome karyotype.He was negative for BCR-ABL(190,210,230),MPL,and CALR but positive for JAK-2V617,with a variance accounted for(VAF)value of 69.6%.The patient was negative for JAK-2 exon 12 mutations.The results of gene sequencing for ASXL1,EZH2,TET2,IDH1/IDH2,SRSF2,and SF3B1 mutations were negative,as were those for TCR and IgH mutations.In a thrombophilia gene panel,the results were as follows: SERPINC1(NM-000488),intron mutation c.41+141G>A(heterozygous)around exon 1;the FII G20210A and FV G1691A mutations were not present.The patient in this case had a β-thalassemia gene CD14-15 heterozygous mutation,a UGT1A1*28 homozygous mutation,and a SERPINC1 EXON1 intron mutation.The family medical history revealed that his father had yellow sclera when tired or when he had a cold.The related genes of the patient were evaluated,and the results showed that the father had a β-thalassemia gene CD14-15 heterozygous mutation.The mother tested negative for the mutation.Both parents had UGT1A1*28 homozygous mutations and SERPINC1 EXON1 intron mutations(Figure 3).However,the patient's parents had no jaundice,gallstones,thrombi,or splenomegaly,and both had no gallbladder attacks.
Imaging examinations
In July 2019,abdominal ultrasound showed splenomegaly(67 mm × 198 mm)and splenic vein thrombosis.Vascular ultrasound at this time showed abnormal thromboses in the portal vein system and portal vein cavernous changes.In addition,hepatic artery velocity was increased at the beginning of the porta hepatis.A thoracic and abdominal aorta CT spiral scan + three-dimensional reconstruction showed no development in the intrahepatic vein,main portal vein,splenic vein,or parts of the superior mesenteric vein,which was considered to indicate thrombosis.Liver and spleen enlargement,peritoneal effusion,diffuse edema between the retroperitoneum and the mesenteric root,and a density similar to that of a metallic needle near the duodenum were observed.The chest X-ray showed no abnormalities.
FlNAL DlAGNOSlS
Primary myelofibrosis with thrombophilia as the first symptom combined with thalassemia and Gilbert syndrome.
TREATMENT
The patient was administered with rivaroxaban tablets 15 mg/d and ruxolitinib tablets 15 mg twice a day.
OUTCOME AND FOLLOW-UP
That s just where it is, sighed the Caliph, whose wings drooped94 in a dejected manner; how do you know she is young and lovely? I call it buying a pig in a poke26
DlSCUSSlON
Hereditary thrombophilia refers to the body having heredity defects in anticoagulant proteins,blood coagulation factors,and fibrinolytic proteins,leading to a higher tendency for thromboembolism.Hereditary AT deficiency is a hereditary thrombophilia and is a rare autosomal incomplete dominant disease.This hereditary deficiency is caused by mutations in the AT coding gene SERPINCI1[1],and clinically,most patients are heterozygous for these mutations.In 1965,Egeberg first reported a case of familial venous thrombosis due to hereditary antithrombin deficiency.Subsequently,research on hereditary antithrombin deficiency was carried out gradually.According to the three studies conducted by Zhu
[2],Ding
[3],and Gu
[4]in China,the proportion of hereditary AT deficiency in the population with venous thrombosis in China is approximately 3.67%(95%confidence interval,2.55%-4.78%).Individuals with hereditary AT deficiency in the Chinese population have a 43-fold increased risk of venous thrombosis(95% confidence interval,13-138)[5].Generally,regarding the role of genetic factors and AT gene mutations,the risk of venous thromboembolism in people with AT deficiency is several times higher than that of patients with no mutation in this gene.However,the population with AT gene mutations will not all develop heredity AT deficiency,and the occurrence of heredity AT deficiency is also affected by acquired risk factors,such as surgery,trauma,long periods of immobilization,advanced age,malignant tumors,oral contraceptives,and pregnancy,and under the dual effect of hereditary genetic mutations and acquired risk factors,the occurrence of heredity AT deficiency will be significantly increased.The patient in this case had a SERPINC1 heterozygous mutation and slightly decreased AT-III activity.In addition,his parents both had a SERPINC1 heterozygous mutation.According to current database research,this mutation was identified as a single-nucleotide polymorphism,but some studies have indicated that it is still a risk factor for venous thrombosis[6-8].Therefore,the diagnosis of a hereditary antithrombin defect was clear.
The patient in this case had the primary symptom of sigmoid sinus and transverse sinus venous thrombosis at the age of 42 years,followed by regular warfarin tablet usage for a long period,and then he developed venous thrombosis again at the age of 44,with a mildly low AT-III activity level.The patient and both his parents had the SERPINC1 mutation,but neither of his parents had a history of thrombosis,so it is difficult to explain the two severe thrombophilic incidents in the patient.The patient was found to have splenomegaly for approximately 2 years,with obvious fatigue and discomfort in the last 3 mo.We suspected that there were other acquired thrombophilia risk factors that aggravated the disease.The leukocyte and PLT counts and HB level of the patient were normal,but MCV and MCHC were low,and the EPO level was normal.A peripheral blood smear showed erythroblasts.A bone marrow smear showed no dyshematopoiesis.Bone marrow biopsy showed multifocal fibrous tissue hyperplasia and focal fibrosis(MF-1)among bone trabeculae.The patient was positive for the JAK-2V617 mutation,with a VAF value of 69.6%,but negative for ASXL1,EZH2,TET2,IDH1/IDH2,SRSF2,SF3B1 TCR,and IgH gene mutations.According to early primary myelofibrosis(pre-PMF),at the early stage of PMF,his diagnostic criteria depended on the histopathological features of bone marrow biopsy,fibrosis grade,clinical parameters(leukocytosis,anemia,rising LDH,and palpable splenomegaly),and other indicators[9].Regardless of whether it is dominant or recessive,PMF itself is a major intrinsic factor in the development of thrombosis.Hereditary thrombophilia is an important risk factor[10-13]for PMF thromboembolism.The positive JAK-2V617 pre-PMF in this patient was a potential secondary risk factor for recurrent venous thrombosis.
The patient had jaundice that preceded his venous thrombosis,and fatigue and cold aggravated his jaundice.Erythrocyte morphology indicated hypochromic microcytic anemia.The RET proportion was mildly increased.Jaundice was a manifestation of an increase in indirect bilirubin.Bone marrow smear and biopsy suggested that erythroid hyperplasia was active.We suspected autoimmune hemolytic anemia and paroxysmal nocturnal hemoglobinuria.However,the results of hemoglobin electrophoresis showed that plasma HbF was increased and HB was slightly reduced,and genetic testing revealed UGT1A1*28 missense mutations and thalassemia gene CD14-15 heterozygous mutations.The patient had a coexisting thalassemia gene CD14-15 heterozygous mutation and UGT1A1 gene mutation,which could be confirmed as a combination of β-thalassemia(mild)and Gilbert syndrome.The patient's father also had a history of intermittent yellow scleral staining,but the parents' spleens were not enlarged,and their routine blood test results and bilirubin levels were normal.Gene sequencing confirmed that his parents both had a UGT1A1*28 missense mutation,and the father also had a thalassemia gene CD14-15 heterozygous mutation,but neither of the parents was affected by the disease.
Mike, seated beside me, shook his head sadly, I wish just one of them could have won, he said. They have a lot of potential, but losing like this could take the heart right out of them.
In conclusion,the patient in this case had thrombophilia as the primary symptom,JAK2V617-positive MPN was the main potential cause,and hereditary AT-III deficiency may have been one of multiple secondary causes.It remains to be determined whether UGT1A1 and β-thalassemia gene mutations are related to thrombophilia.However,the clinical features of MPN in this patient were hidden,and the relevant clinical features of coexisting thalassemia and hereditary Gilbert syndrome,reported here for the first time domestically and abroad,were complicating factors,causing great difficulties for a clear diagnosis.Thus,when thrombophilia has been determined,it is necessary to screen the relevant latent problems overall.When the clinical features cannot be perfectly explained by one etiology,a relevant comprehensive examination should also be initiated from the perspective of multiple etiologies.
The results of routine blood tests were as follows: WBCs,3.82 × 10
/L;HB,121 g/L[mean corpuscular volume(MCV),65.9 fl;mean corpuscular hemoglobin(MCH),22.2 pg;mean corpuscular hemoglobin concentration(MCHC),314 g/L];PLTs,301 × 10
/L;RETs,3.77%;and absolute RET count,85.86 × 10
/L.The results of a biochemistry panel were as follows: Total bilirubin,70.8 μmol/L;direct bilirubin,9.99 μmol/L;indirect bilirubin,60.81 μmol/L;lactate dehydrogenase(LDH),184 U/L;erythropoietin(EPO),3.41 mIU/mL;ferritin,and 145 ng/mL.The result of an immunofixation electrophoresis test was negative.The results of tests for antinuclear antibody(ANA),double-stranded DNA(dsDNA),extractable nuclear antibody(ENA),and antineutrophil cytoplasmic antibody(ANCA)were all negative.Test results for antibodies associated with autoimmune liver disease and liver fibrosis were negative.Coagulation function test results were as follows: Prothrombin time(PT),19.1 s;PT to international normalized ratio(INR)ratio;INR,1.72;fibrinogen(FIB),2.08 g/L;activated partial thromboplastin time(APTT),48.3 s;D-dimer,0.49 mg/L;AT-III,53.8%(normal range,75%-125%);protein C-1,81%(70-140);protein S,81%(55-140);coagulation factor 8,42%;coagulation factor 9,45%;von Willebrand factor,81(50-160);and activated protein C resistance test,283.6(120-300)s.Anticardiolipin antibody was < 2 RU/mL(< 12),lupus anticoagulant was negative,and thromboelastography was normal.A clot retraction test showed complete retraction.The activity of internal and external coagulation factors was normal.The results of an antiglobulin test,Ham test,acidified hemolysis,CD55,CD59,and Flear were negative.Abdominal ultrasound showed intrahepatic portal vein plugging and collateral angiogenesis.
The results of routine blood tests were as follows: White blood cells(WBCs),4.1 × 10
/L;hemoglobin(HB),126 g/L;platelets(PLTs),259 × 10
/L;reticulocytes(RETs),3.77%;total bilirubin,59.5 μmol/L;and indirect bilirubin,50.53 μmol/L.
CONCLUSlON
A healthy person has two β globin genes,with one on each chromosome.Mutations in one or two of these genes can lead to a decrease in expression(β+)or complete deficiency(β
),resulting in βthalassemia.Disease severity is associated with normal β globin production,presenting as small-cell homogeneous hemolytic anemia,which is also one of the common single-gene genetic diseases in humans[14],and genetic diagnosis is regarded as the "gold standard" for this disease.Also known as βthalassemia carriers,patients with mild β-thalassemia are characterized by a β+ or β0 thalassemia mutational heterozygous carrying state.Mild β-thalassemia is often an asymptomatic carrier condition.Patients may be mildly anemic and most are asymptomatic but may have microcytosis that may be mistaken for iron deficiency.This patient showed fatigue but no anemia,while the cell shape was microcytic,which can be diagnosed as mild β-thalassemia.A retrospective study in 2008 included 217 patients with β-thalassemia characteristics to explore the possibility of other mild symptoms or test findings[15].The clinical presentation of the patient and his parents was consistent with that in this study.About two-thirds of patients knew their diagnosis.The control group included 89 healthy persons and 96 patients with microcyte anemia due to other causes(such as mild iron deficiency).Compared with the control group,patients with β-thalassemia characteristics were more likely to have symptoms of anemia(lethargy and fatigue)and more likely to see doctors for fever.The symptoms of patients with β-thalassemia characteristics were similar to those of patients with other causes of microcytic anemia.In mild thalassemia,the spleen is not palpable,but abnormal enlargement of the spleen can be seen by ultrasound[15].The spleen may progressively enlarge,leading to early satiety.
FOOTNOTES
Please don t touch it, cried Whitey in great distress9. The cabbages are the walls of my house, and if you eat them you will make a hole, and the wind and rain will come in and give me a cold. Do go away; I am sure you are not a friend, but our wicked enemy the fox. And poor Whitey began to whine18 and to whimper, and to wish that she had not been such a greedy little pig, and had chosen a more solid material than cabbages for her house. But it was too late now, and in another minute the fox had eaten his way through the cabbage walls, and had caught the trembling, shivering Whitey, and carried her off to his den.
Informed written consent was obtained from the patient for publication of this report and any accompanying images.
Wufuer G has received fees for serving as a speaker,an Associate Chief Physician at People's Hospital of Xinjiang Uygur Autonomous Region;Duan MH has received research funding from Peking Union Medical College Hospital.
So it went for the next couple of months, as Justin checked every detail of his precious plum tree and reported to me about the flowers turning to tiny beads6 that would become plums.
After the diagnosis,the patient did not agree to pharmacological intervention for myelofibrosis,and he continued to take 20 mg of rivaroxaban tablets outside of the hospital once a day for anticoagulant therapy.He lost 10 kg of weight in the last year,and his fatigue and discomfort were significantly increased,accompanied by systemic bone pain but no fever or night sweats.He was followed at our hospital again in April 2021.His routine blood examination results were as follows: WBCs,4.94 × 10
/L;HB,159 g/L(MCV,60.9 fl;MCH,20.6 pg;MCHC,333 g/L);PLTs,301 × 10
/L;neutrophils,3.49 × 10
/L;and lymphocytes,1.13 × 10
/L.Abdominal ultrasound showed wall thickening and echo enhancement of the intrahepatic Gleason system.A lack of echo in the tortuous tubules could be seen around the main portal vein and its branches,one of which had an inner diameter of approximately 1.1 cm,and striped hypoechoic filling could be seen in the lesion part of the left portal vein.A visible tubular anechoic vein extended from the left branch of the portal vein to the body surface.The width of the wider tubular anechoic vein was approximately 0.7 cm.The spleen thickness was approximately 9.2 cm,and the subcostal thickness was approximately 9.9 cm.The capsule was smooth,and the solid echo was uniform.Color Doppler flow imaging showed no blood flow signal in the lesion part of the left portal vein.The velocity of one portal vein was approximately 14 cm/s.A diagnosis of cavernous degeneration of the portal vein,wall thickening and echo enhancement of the intrahepatic Glisson system,umbilical vein opening,a thrombus in the lesion part of the left portal vein,and splenomegaly was made.No obvious abnormalities were found in the bone marrow granulocyte or erythrocyte system after reexamination.More porous and intersected reticular fibers were found in bone trabeculae,with a fibrosis grade of MF-1.The patient was positive for the JAK-2 V617F gene mutation,and the VAF value was 73.3%.On May 10,2021,the patient began to take ruxolitinib tablets 5 mg twice a day,and he felt that his abdominal distension was relieved after 2 wk of taking the tablets.Currently,the dosage of ruxolitinib tablets is 15 mg twice a day,which the patient can tolerate,and his PLT count is above 150 ×10
/L.HB was maintained above 120 g/L.He denied dizziness,headache,abnormal liver function,and other adverse conditions.
The authors have read the CARE Checklist(2016),and the manuscript was prepared and revised according to the CARE Checklist(2016).
As I cared for my patients, George was right alongside. I watched him spread holiday cheer as he became a guest to the patients who had no visitors that day. When trays arrived he knew who needed assistance and who needed to be fed. He read letters and cards to those whose eyes could no longer see the letters on a printed page. George’s powerful body and tender hands were always ready to help hold, turn, pull-up or lift a patient. He was a “gopher” who made countless15 trips to the supply room for the “needs of the moment.”
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See: http://creativecommons.org/Licenses/by-nc/4.0/
I ran as fast as I could back through the orphanage gate and into the thick azalea bushes. I uncovered my home-made bow which I had constructed out of bamboo and string. I grabbed four arrows that were also made of bamboo and they had coca cola tops bent4 around the ends to make real sharp tips. Then I ran back out the gate with an arrow cocked in the bow and I just stood there quiet like, breathing real hard just daring either one of them to kick or touch the boy again.
China
Guzailinuer Wufuer 0000-0001-7423-2929;Kaisaer Wufuer 0000-0002-3099-5235;Tu Ba 0000-0003-2581-7589;Tao Cui 0000-0003-3538-991X;Ling Tao 0000-0002-3675-4918;Ling Fu 0000-0002-2276-1236;Ming Mao 0000-0002-4952-277X;Ming-Hui Duan 0000-0003-2130-6300.
Yan JP
Wang TQ
Yan JP
1 Bock SC,Wion KL,Vehar GA,Lawn RM.Cloning and expression of the cDNA for human antithrombin III.
1982;10: 8113-8125[PMID: 6298709 DOI: 10.1093/nar/10.24.8113]
2 Zhu T,Ding Q,Bai X,Wang X,Kaguelidou F,Alberti C,Wei X,Hua B,Yang R,Wang Z,Ruan C,Schlegel N,Zhao Y.Normal ranges and genetic variants of antithrombin,protein C and protein S in the general Chinese population.Results of the Chinese Hemostasis Investigation on Natural Anticoagulants Study I Group.
2011;96: 1033-1040[PMID: 21486865 DOI: 10.3324/haematol.2010.037515]
3 Ding Q,Wang M,Xu G,Ye X,Xi X,Yu T,Wang X,Wang H.Molecular basis and thrombotic manifestations of antithrombin deficiency in 15 unrelated Chinese patients.
2013;132: 367-373[PMID: 23932013 DOI:10.1016/j.thromres.2013.07.013]
4 Gu Y,Shen W,Zhang L,Zhang J,Ying C.Deficiency of antithrombin and protein C gene in 202 Chinese venous thromboembolism patients.
2014;36: 151-155[PMID: 24028705 DOI: 10.1111/ijlh.12146]
5 Tang L,Hu Y.Ethnic diversity in the genetics of venous thromboembolism.
2015;114: 901-909[PMID:26156046 DOI: 10.1160/TH15-04-0330]
6 Austin H,De Staercke C,Lally C,Bezemer ID,Rosendaal FR,Hooper WC.New gene variants associated with venous thrombosis:A replication study in White and Black Americans.
2011;9: 489-495[PMID: 21232005 DOI: 10.1111/j.1538-7836.2011.04185.x]
7 Antón AI,Teruel R,Corral J,Miñano A,Martínez-Martínez I,Ordóñez A,Vicente V,Sánchez-Vega B.Functional consequences of the prothrombotic SERPINC1 rs2227589 polymorphism on antithrombin levels.
2009;94:589-592[PMID: 19229049 DOI: 10.3324/haematol.2008.000604]
8 Yue Y,Sun Q,Xiao L,Liu S,Huang Q,Wang M,Huo M,Yang M,Fu Y.Association of
Gene Polymorphism(rs2227589)With Pulmonary Embolism Risk in a Chinese Population.
2019;10: 844[PMID: 31572449 DOI:10.3389/fgene.2019.00844]
9 Arber DA,Orazi A,Hasserjian R,Thiele J,Borowitz MJ,Le Beau MM,Bloomfield CD,Cazzola M,Vardiman JW.The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia.
2016;127:2391-2405[PMID: 27069254 DOI: 10.1182/blood-2016-03-643544]
10 Mishcheniuk OY,Kostukevich OM,Dmytrenko IV,Sholoyko VV,Prokopenko IM,Martina ZV,Pilipenko GV,Klymenko SV.Molecular characterization of Ph-negative myeloproliferative neoplasms in Ukraine.
2013;35:202-206[PMID: 24084459]
11 Mishcheniuk OY,Shkarupa VM,Kostukevich OM,Neumerzhitcka LV,Kravchenko SM,Klymenko SV.The contribution of hereditary thrombophilia to increasing the frequency of thrombosis in patients with Ph negative myeloproliferative neoplasms,including the victims from the Chornobyl accident.
2016;21: 291-311[PMID:28027559]
12 Yildiz A,Güryildirim M,Pepeler MS,Yazol M,Oktar SÖ,Acar K.Assessment of Endothelial Dysfunction With Flow-Mediated Dilatation in Myeloproliferative Disorders.
2018;24: 1102-1108[PMID: 29683036 DOI: 10.1177/1076029618766260]
13 Horvat I,Boban A,Zadro R,Antolic MR,Serventi-Seiwerth R,Roncevic P,Radman I,Sertic D,Vodanovic M,Pulanic D,Basic-Kinda S,Durakovic N,Zupancic-Salek S,Vrhovac R,Aurer I,Nemet D,Labar B.Influence of Blood Count,Cardiovascular Risks,Inherited Thrombophilia,and JAK2 V617F Burden Allele on Type of Thrombosis in Patients With Philadelphia Chromosome Negative Myeloproliferative Neoplasms.
2019;19: 53-63[PMID: 30301673 DOI: 10.1016/j.clml.2018.08.020]
14 Cappellini MD,Cohen A,Eleftheriou A,Piga A,Porter J,Taher A.Guidelines for the Clinical Management of Thalassaemia[Internet].Nicosia(CY): Thalassaemia International Federation;2008[PMID: 24308075]
15 Schmid R.Gilbert's syndrome--a legitimate genetic anomaly?
1995;333: 1217-1218[PMID: 7565981 DOI:10.1056/NEJM199511023331812]
16 Muraca M,Fevery J.Influence of sex and sex steroids on bilirubin uridine diphosphate-glucuronosyltransferase activity of rat liver.
1984;87: 308-313[PMID: 6428963]
17 Powell LW,Hemingway E,Billing BH,Sherlock S.Idiopathic unconjugated hyperbilirubinemia(Gilbert's syndrome).A study of 42 families.
1967;277: 1108-1112[PMID: 6054997 DOI: 10.1056/NEJM196711232772102]
18 Ellis E,Wagner M,Lammert F,Nemeth A,Gumhold J,Strassburg CP,Kylander C,Katsika D,Trauner M,Einarsson C,Marschall HU.Successful treatment of severe unconjugated hyperbilirubinemia
induction of UGT1A1 by rifampicin.
2006;44: 243-245[PMID: 16288819 DOI: 10.1016/j.jhep.2005.09.011]
19 Lee HJ,Moon HS,Lee ES,Kim SH,Sung JK,Lee BS,Jeong HY,Lee HY,Eu YJ.A case of concomitant Gilbert's syndrome and hereditary spherocytosis.
2010;16: 321-324[PMID: 20924216 DOI:10.3350/kjhep.2010.16.3.321]
20 Jiang J,Wang HG,Wu WL,Peng XX.Mixed Dubin-Gilbert Syndrome:A Compound Heterozygous Phenotype of Two Novel Variants in
Gene.
2017;130: 1003-1005[PMID: 28397734 DOI:10.4103/0366-6999.204108]
21 Li XX,Shi J,Huang ZD,Shao YQ,Nie N,Zhang J,Ge ML,Huang JB,Zheng YZ.[Clinical Characteristics and Gene Mutations of Gilbert Syndrome Complicated with Myeloproliferative Neoplasm].
2017;25: 567-571[PMID: 28446312 DOI: 10.7534/j.issn.1009-2137.2017.02.046]
22 Engelborghs S,Pickut BA,De Deyn PP.Recurrent transient ischemic attacks in a 15-year-old boy with beta-thalassemia minor and thrombophilia.Contribution of perfusion SPECT to clinical diagnosis.
2003;103: 99-102[PMID: 12892004 DOI: 10.1007/s00701-003-0019-0]
杂志排行

World Journal of Clinical Cases的其它文章
- Capillary leak syndrome:A rare cause of acute respiratory distress syndrome
- lmproving outcomes in geriatric surgery:ls there more to the equation?
- Mass brain tissue lost after decompressive craniectomy:A case report
- Primary intracranial extraskeletal myxoid chondrosarcoma:A case report and review of literature
- Spinal canal decompression for hypertrophic neuropathy of the cauda equina with chronic inflammatory demyelinating polyradiculoneuropathy:A case report
- Enigmatic rapid organization of subdural hematoma in a patient with epilepsy:A case report