海南岛淡水鱼类环境DNA宏条形码参考数据库的初步构建及比较分析
2022-06-22蔡杏伟张清凤李高俊马春来申志新
陈 治,蔡杏伟,张清凤,李高俊,马春来,申志新
1. 海南热带海洋学院/热带海洋生物资源利用与保护教育部重点实验室/海南省热带海洋渔业资源保护与利用重点实验室,海南 三亚 572022
2. 海南省海洋与渔业科学院,海南 海口 571126
鱼类是淡水生态系统的重要组成部分。目前,有超过10 000种鱼类生活在淡水中,大约占鱼类总数的40%和脊椎动物的1/4[1]。然而,由于气候变化、人类活动、生物入侵等原因,全球淡水鱼类正以前所未有的速度消失。淡水鱼类保护成为当前生物多样性保护刻不容缓的事宜[1]。全面准确的多样性调查是开展各项淡水鱼类保护的基础。传统的淡水鱼类调查一般基于网具捕获 (如网捕、笼捕等),这对调查对象及其所在生境具有一定破坏性。随着分子生物学技术的发展,一种对调查对象无损伤、对环境友好的物种多样性调查方法——环境 DNA (Environmental DNA, eDNA) 宏条形码(Metabarcoding) 技术应运而生[2]。该技术是指对从环境样品 (如底泥、水、粪便等) 中分离的总DNA进行目的片段的PCR扩增和高通量测序,通过与已有DNA数据库进行比对与注释,从而实现多物种 (或更高级分类单元) 的鉴定[3]。近年来,环境DNA宏条形码技术引起了渔业生态学家的广泛关注,并逐渐应用于淡水鱼类多样性调查、珍稀濒危物种和外来入侵种检测等领域[4-6]。
然而,作为一种新兴的水生生物多样性调查方法,环境DNA宏条形码技术目前仍存在很多问题——不仅取样策略、实验环境对研究结果具有较大影响[2,7],而且物种判别方案对定性、定量的准确性也有不可忽视的影响[2-3,7]。目前,物种判别方面比较突出的问题是:1) 参考数据库的构建。既可以选择NCBI、BOLD、MitoFish、FISH-BOL等公共数据库,也可以实际采集样品自建数据库,或者二者相结合[7]。主流的观点认为自建数据库优于公共数据库[8-9],但目前大部分鱼类环境DNA宏条形码研究主要还是采用公共数据库[10-11],且其中一些研究表明基于公共数据库进行物种注释效果也比较理想[12-14]。因此数据库的优劣可能跟研究区域、物种类群等因素密切相关。2) 最优目标基因的选择。目前已经有不少针对鱼类设计的环境DNA宏条形码通用引物[7,15-16],其目标基因各不相同,对鱼类的鉴定能力也存在差异。究竟哪种目标基因更适合特定的研究区域,需要根据具体调查类群进行筛选[7,15-16]。3) 种间差异阈值的确定。标准的动物DNA条形码主要为线粒体细胞色素c氧化酶亚基I(Cytochrome coxidase subunit I, COI) 基因;但鱼类环境DNA 宏条形码主要选用线粒体12S核糖体(12s ribosomal RNA, 12S) 或 16S 核糖体 (16s ribosomal RNA, 16S) 基因[16]。不仅变异速度更不稳定[17],且扩增片段多小于200 bp[15-16]。物种系统发育分析过程的种间差异阈值应为多少目前也尚无定论[18]。
海南省是中国唯一的热带岛屿省份,气候条件优越,是中国生物多样性的天然宝库和资源基地,有着重要的保护价值[19]。2016—2018年,海南省海洋与渔业科学院对本省淡水鱼类进行了比较全面的调查[20],至少采集到淡水鱼类124种 (不包括2种洄游的鳗鲡)——仅“两江一河” (南渡江、昌化江、万泉河) 初步确认的淡水土著鱼类就达93种[21],其中海南岛特有鱼类19种[21]。考虑到传统调查方法具有费时费力、破坏性大及靶生物捕获率低的缺点,可能还有更多的土著种及特有种等待发掘。有必要在后续调查研究中引入环境DNA宏条形码技术,从而更好地了解海南岛土著鱼类多样性。而参考数据库的准确构建和条形码基因的合理选择,则是后续使用该技术的基础和前提。针对海南岛淡水鱼类多样性调查的实际需求及环境DNA宏条形码技术存在的问题,本研究的目的如下:1) 初步构建海南岛淡水鱼类环境DNA宏条形码参考数据库,比较自建数据库与公共数据库在物种注释上的差异;2) 以自建数据库为本底资料,考察不同鱼类的种间差异,探究不同目标基因的物种判别能力及种间差异阈值。本研究将为后期基于环境DNA宏条形码技术的海南岛淡水鱼类多样性调查和其他类似研究提供基础和参考。
1 材料与方法
1.1 样品采集及鉴定
样品采集及鉴定由海南省海洋与渔业科学院完成。采集时间始于2016年,采集地点为海南岛各淡水水系。形态鉴定主要参照《海南岛淡水及河口鱼类志》[22]《广东淡水鱼类志》[23]等资料。对于形态鉴定无误的物种,剪取偶鳍鳍条或背部组织肌肉,于无水乙醇中−20 ℃保存。
1.2 条形码获取
DNA提取采用标准的苯酚-氯仿-异戊醇法。使用目前应用广泛的鱼类环境DNA宏条形码通用引物MiFish-U[17](针对线粒体12S,扩增子约170 bp)、Vert-16S[24](针对线粒体 16S,扩增子约 256 bp)及本研究基于148种鱼类的COI序列专门针对海南岛淡水鱼类设计的COI短片段引物 (F: AAYCAYAAAGACATYGGYACCCT,R: GGYATTACTATAAAGAARATYAT,扩增子 139 bp) 进行PCR扩增。PCR产物及后续处理参照吴娜[25]、梁日深等[26]:反应体系总体积为50 μL,其中包括PCR Mix 反应混合液 (天根生化科技有限公司) 25 μL、灭菌蒸馏水 21 μL、上下游引物 (10 μmol·L−1)各 1 μL、DNA 样品 2 μL。PCR 反应条件为 94 ℃预变性 5 min;94 ℃ 变性 30 s,55 ℃ 退火 30 s,72 ℃ 延伸 40 s,35 个循环;最后 72 ℃ 再延伸5 min。PCR产物用1%琼脂糖凝胶电泳检测,纯化回收后送广州艾基生物技术有限公司进行双向测序。此外,对于部分有历史分布记录但实际暂未采集到相关样品的鱼类,或者由于样品个体很小、状态保存差而测序失败的疑似种,本研究一律按该物种在海南有分布处理。从NCBI公共数据库 (以下简称公共数据库) 下载这些物种的线粒体序列,截齐后暂时作为海南岛淡水鱼类环境DNA宏条形码参考序列 (具体名单及GenBank序列号见附录A,详见 http://dx.doi.org/10.12131/20210339 的资源附件)。
1.3 序列注释
基于序列相似度,模拟条形码序列注释过程,统计测序所得序列在公共数据库和自建数据库的物种注释情况。待注释序列在参考数据库中比对到的高相似度物种有且仅有其自身1种时,则表示该序列被准确注释。基于公共数据库的序列注释操作如下:打开NCBI序列比对窗口 (https://blast.ncbi.nlm.nih.gov/Blast.cgi),选择“Nucleotide Blast”,将人工校对切齐处理后的序列输入“Enter accession number (s), gi (s), or FASTA sequence (s)”对话框,点击“Blast”选项统计不同相似度下的物种比对名录;基于自建数据库的注释过程则参见郜星晨和姜伟[27]:安装BLAST-2.4软件工具并配置系统工作环境,将COI、12S、16S序列整合为3个不同的FASTA格式文件 (COI-barcode.fa、12S-barcode.fa和16S-barcode.fa),Makeblastdb命令格式化和索引化上述数据 (参数:-in COI-barcode.fa -dbtype nucl -parse_seqids -out fish、-in 12S-barcode.fa -dbtype nucl -parse_seqids -out fish、-in 16S-barcode.fa -dbtype nucl -parse_seqids -out fish),初步形成可供BLAST检索的本地数据库。取待检测物种的序列,保存为test.fa文件。Blastn命令执行待检序列数据库检索 (参数:-query test.fa -db fish -evalue 1e-5-outfmt 2),自动输出检索结果 test.txt。
1.4 系统发育及阈值分析
邻接系统发育 (Neighbor-joining, NJ) 分析、种间差异阈值的调整及确定参考Milan等[18]。具体操作如下:测定的序列通过DNAStar软件包中的Seqman程序进行人工校对切齐。打开MEGA 6.0软件,基于Kimura 双参数模型 (Kimura-2-parameter,K2P) 采用邻接法构建系统发育树。系统分支支持率经1 000次重复抽样检测;统计序列间的遗传距离,在0~0.02范围内以0.000 5为阈值间隔设置不同的种间差异阈值,物种错误鉴定比例最小的阈值则为种间差异最佳阈值。
2 结果
2.1 数据库简介
截至2021年5月1日,本研究实地采集鱼类72种 (共85尾,其中12种鱼类样品量为2~3尾,其余种类样品量仅1尾) ,从公共数据库下载67种 (附录A)。两数据库物种共计139种,隶属于8目25科95属 (附录A)。其中鲤形目、鲈形目和鲇形目物种数较多,分别为73、37和18种;鱂形目、脂鲤目、颌针鱼目、合鳃鱼目和鳗鲡目数量较少,种类仅2~3种。
虽然本次建库实地采集的鱼类种数还较少,但其中却包含南渡江吻虾虎鱼(Rhinogobius nandujiangensis)、海南原缨口鳅 (Vanmanenia hainanensis)等海南岛淡水土著鱼类特有种11种 (具体名单见附录 A)。72 种鱼中,有 16 (COI)、20 (12S) 和 22(16S) 种鱼类在公共数据库内无参考序列,为本研究首次提供 [海南异鱲 (Parazacco fasciatus)、海南华鳊 (Sinibrama melrosei) 等,主要为海南岛淡水土著鱼类特有种或少见种 (附录A)]。
2.2 物种覆盖度
自建数据库有日本鳗鲡 (Anguilla japonica)、中华沙塘鳢 (Odontobutis sinensis) 等 67 种鱼类暂未采到样品,物种覆盖度为51.80% (72/139);公共数据库有 16 (COI)、22 (12S) 、24 (16S) 种鱼类在其学名下无对应序列,物种覆盖度分别为88.49% (COI:123/139)、84.17% (12S: 117/139) 和 82.73% (16S:115/139) (图1、附录A)。自建数据库的物种覆盖度低于公共数据库。
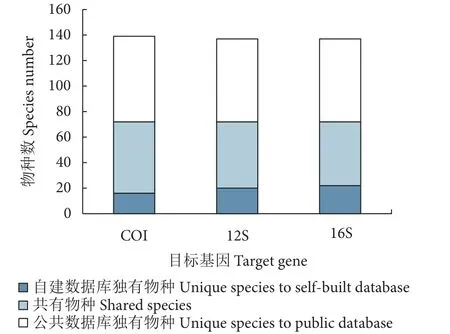
图1 本研究中自建数据库及公共数据库的鱼类种数Fig. 1 Number of fish species in self-built database and public database in this study
公共数据库内的参考序列总数远高于自建数据库的序列总数,但前者明确标注采样地点的序列总数较少,占比分别为6.69% (COI)、11.57% (12S)和8.02% (16S) (表1)。特别是样品采集于海南岛的序列总数和鱼类种数严重不足,占标注采样地点的鱼类种数和序列总数的0~4.42%。而自建数据库72种鱼类皆有详细的采样地点信息和对应的实物样品。此外,公共数据库内不同条形码参考序列数量差别明显。线粒体12S、16S 条形码的序列总数明显低于COI,分别只有后者的23.96%和27.22%。表明公共数据库不同条形码数据库的完善程度也存在较大差异。
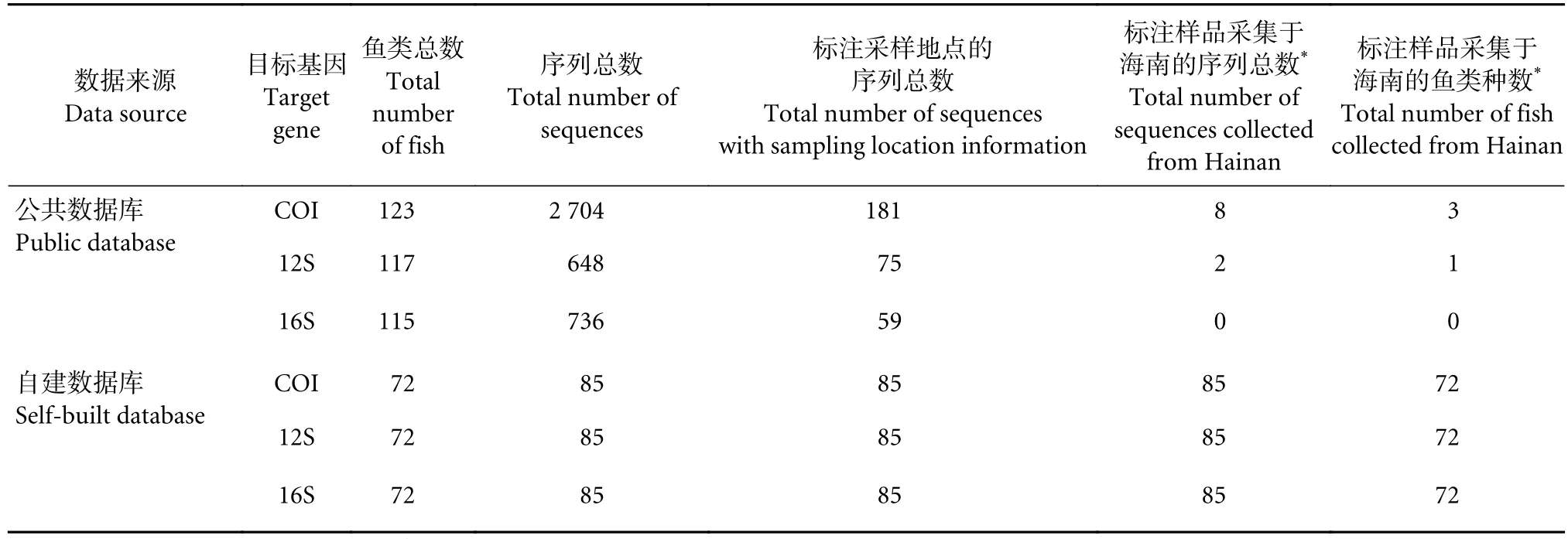
表1 公共数据库和自建数据库参考序列简介Table 1 Summary of metabarcoding reference sequence in public database and self-built database
2.3 注释过程中的候选物种数
自建数据库的物种注释结果显示:72种鱼中,有 23 (COI)、24 (12S) 、22 (16S) 种鱼类在公共数据库内比对不到高相似度序列 (序列相似度<98%),属于不可注释到种的序列,只能大致推测其隶属的科或属。只有 68.06% (COI: 49/72)、66.67% (12S: 48/72) 和 69.44% (16S: 50/72) 的鱼类顺利比对到高相似度序列 (序列相似度≥98%),属于可注释到种的序列。然而,与可注释到种的序列高度相似的物种较多,即使将序列比对阈值提高到≥99%,平均每种序列仍有 2.47 (COI)、1.53 (12S) 、1.85 (16S) 种候选物种 (图2、表2)。候选物种数大于1表明数据库内存在不同鱼类的参考序列高度相似或完全相同现象。而上述可注释到种的序列基于自建数据库都能比对到100%相似度的鱼类,且每种序列的候选物种数明显更少,分别只有1.14(COI)、1.10 (12S) 、1.09 (16S) 种 (取序列相似度≥99%) (图2、表2)。基于两个数据库的候选物种数存在显著或极显著差别 (COI:F=18.93,P<0.000 1;12S:F=4.80,P=0.029; 16S:F=12.87,P=0.000 4) (取序列相似度≥99%)。
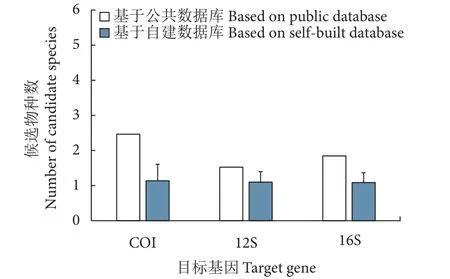
图2 可注释到种的鱼类的候选物种数 (序列相似度≥99%)Fig. 2 Number of candidate species of fish that can be annotated at species level (with≥99% sequence similarity)
表2 可注释到种的序列在不同阈值范围内的候选物种数 ()Table 2 Number of candidate species of sequence that can be annotated at species level within different threshold values
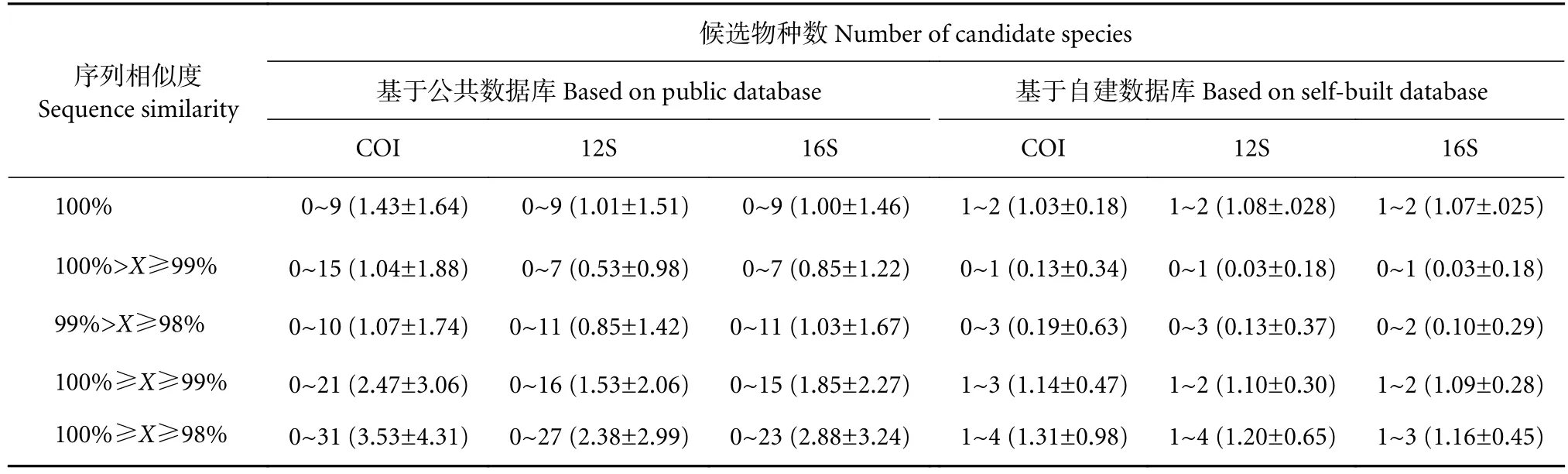
表2 可注释到种的序列在不同阈值范围内的候选物种数 ()Table 2 Number of candidate species of sequence that can be annotated at species level within different threshold values
序列相似度Sequence similarity候选物种数 Number of candidate species基于公共数据库 Based on public database 基于自建数据库 Based on self-built database COI 12S 16S COI 12S 16S 100% 0~9 (1.43±1.64) 0~9 (1.01±1.51) 0~9 (1.00±1.46) 1~2 (1.03±0.18) 1~2 (1.08±.028) 1~2 (1.07±.025)100%>X≥99% 0~15 (1.04±1.88) 0~7 (0.53±0.98) 0~7 (0.85±1.22) 0~1 (0.13±0.34) 0~1 (0.03±0.18) 0~1 (0.03±0.18)99%>X≥98% 0~10 (1.07±1.74) 0~11 (0.85±1.42) 0~11 (1.03±1.67) 0~3 (0.19±0.63) 0~3 (0.13±0.37) 0~2 (0.10±0.29)100%≥X≥99% 0~21 (2.47±3.06) 0~16 (1.53±2.06) 0~15 (1.85±2.27) 1~3 (1.14±0.47) 1~2 (1.10±0.30) 1~2 (1.09±0.28)100%≥X≥98% 0~31 (3.53±4.31) 0~27 (2.38±2.99) 0~23 (2.88±3.24) 1~4 (1.31±0.98) 1~4 (1.20±0.65) 1~3 (1.16±0.45)
不同条形码基因在公共数据库内注释到的候选物种数也存在差别 (表2) ,COI基因高于12S、16S。特别是100%≥X≥99% 范围内三者存在显著差别 (F=4.14,P=0.017) ,表明基于 COI基因的物种注释需要排除更多的物种。
2.4 注释准确率
以序列相似度为主要参考依据,结合Fish-Base、台湾鱼类资料库地理分布记录及《海南岛淡水及河口鱼类志》《广东淡水鱼类志》等,以公共数据库和自建数据库的共有鱼类的测序所得序列为待注释对象,使用两种数据库进行序列注释:基于自建数据库的注释准确率为100% (COI)、96.15%(12S) 和96% (16S);基于公共数据库的物种注释准确率为 69.64% (COI)、67.30% (12S) 和 70% (16S)(表3、附录A)。50~56种共有物种中有11~14种鱼类在公共数据库种比对不到高相似度序列 (表3) ,这直接导致了基于公共数据库的物种注释准确率偏低。
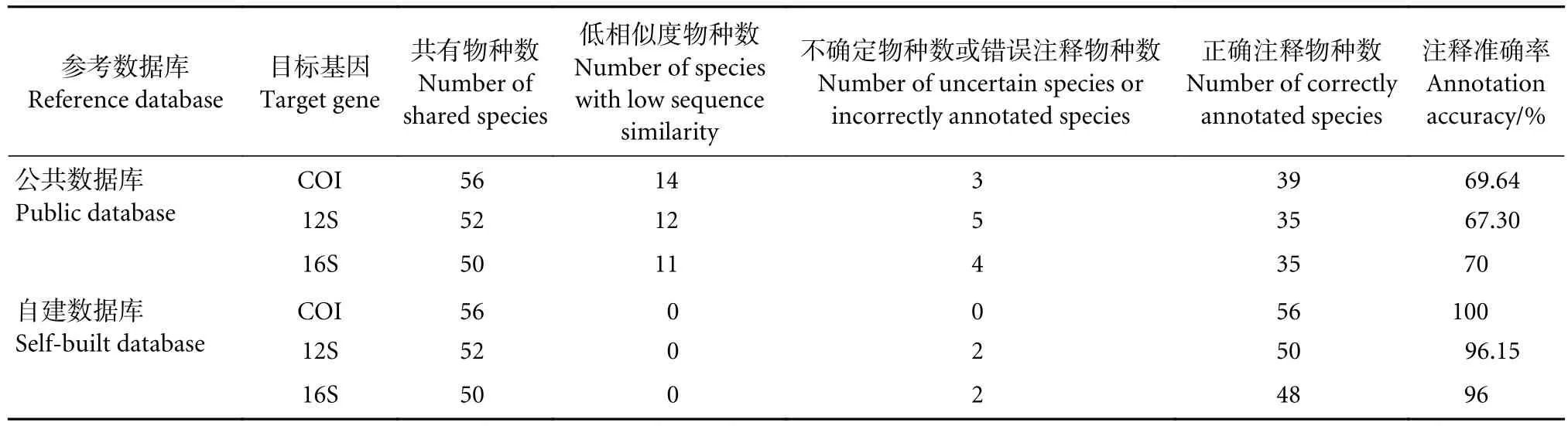
表3 两种数据库共有物种的注释结果Table 3 Annotation results of common fish species in two databases
2.5 系统发育及种间差异阈值分析
基于全部物种的150~152条序列构建的系统进化树见图3—图5。全部序列的总平均遗传距离(Overall mean distance) 分别为 0.206 4 (COI)、0.273 8(12S) 和 0.295 0 (16S)。3 种宏条形码均存在对部分鱼类区分度不够的现象,分别有4 (COI)、12(12S) 、8 (16S) 种鱼类出现种间遗传距离为 0 的情况;而南方马口鱼 (Opsariichthys bidens) 等物种却又出现了种内不同个体遗传差异较大的现象 (表4、图3—图5)。参考Milan等[18]的研究,基于K2P遗传距离确定的种间差异最佳阈值分别为0.006 9(COI)、0.005 6 (12S) 和 0.007 5 (16S) ,其物种判别准确率分别为 94.96% (COI: 132/139)、89.05% (12S:122/137) 和 92.70% (12S: 127/137)。
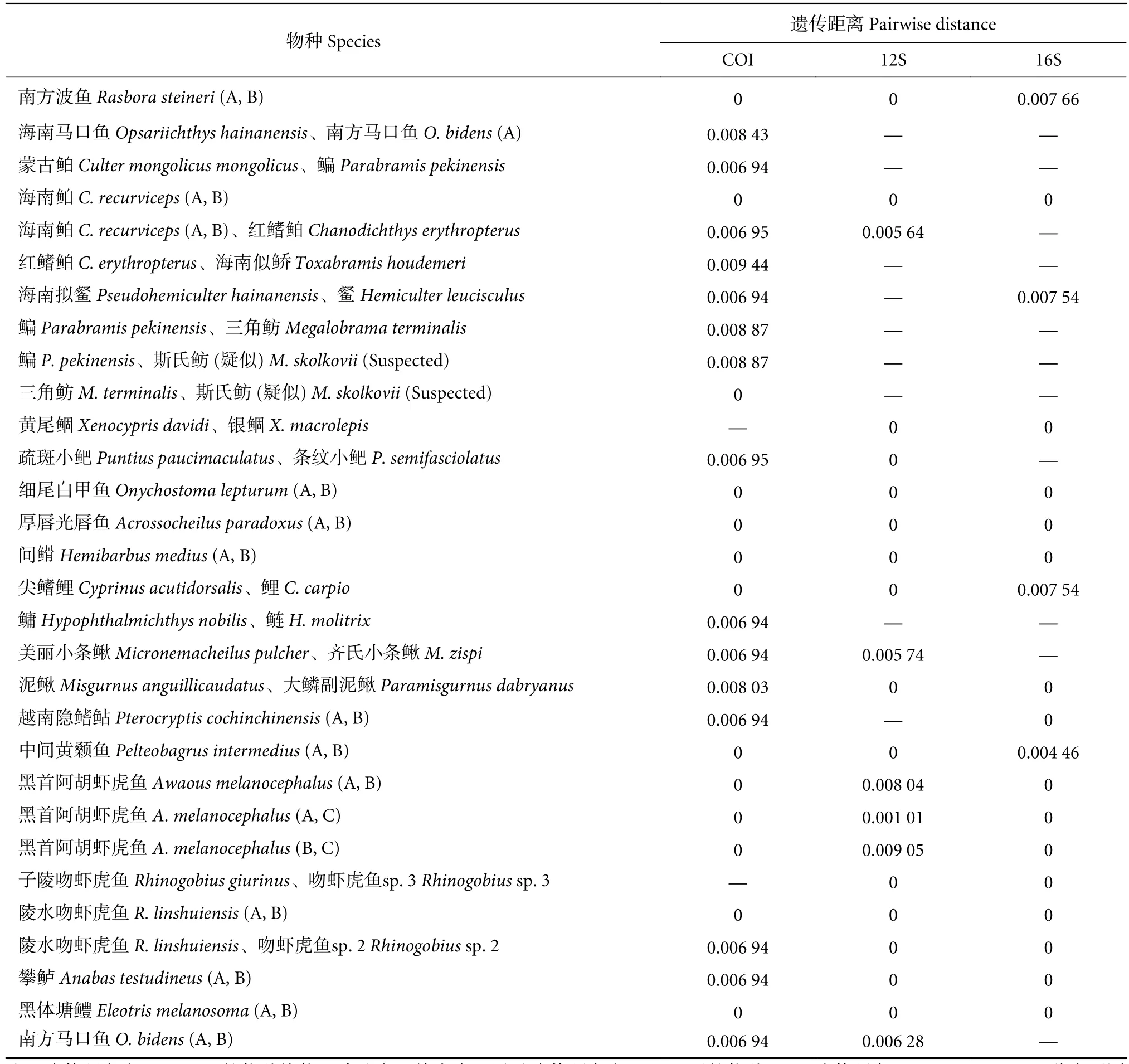
表4 两两序列的遗传距离 (K2P)Table 4 Pairwise distance of genetic divergences (K2P) within various sequences

图3 基于152条线粒体COI序列构建的NJ系统发育树Fig. 3 NJ phylogenetic tree constructed based on 152 mitochondrial COI sequences
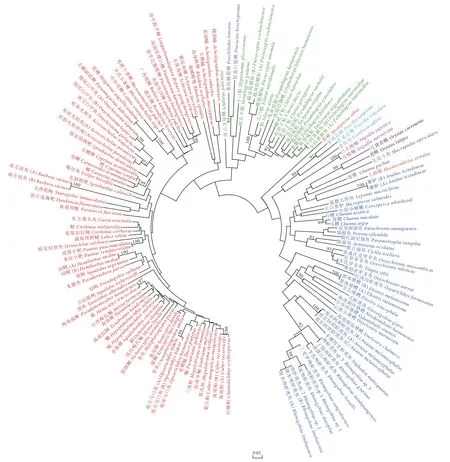
图4 基于150条线粒体12S序列构建的NJ系统发育树Fig. 4 NJ phylogenetic tree constructed based on 150 mitochondrial 12S sequences
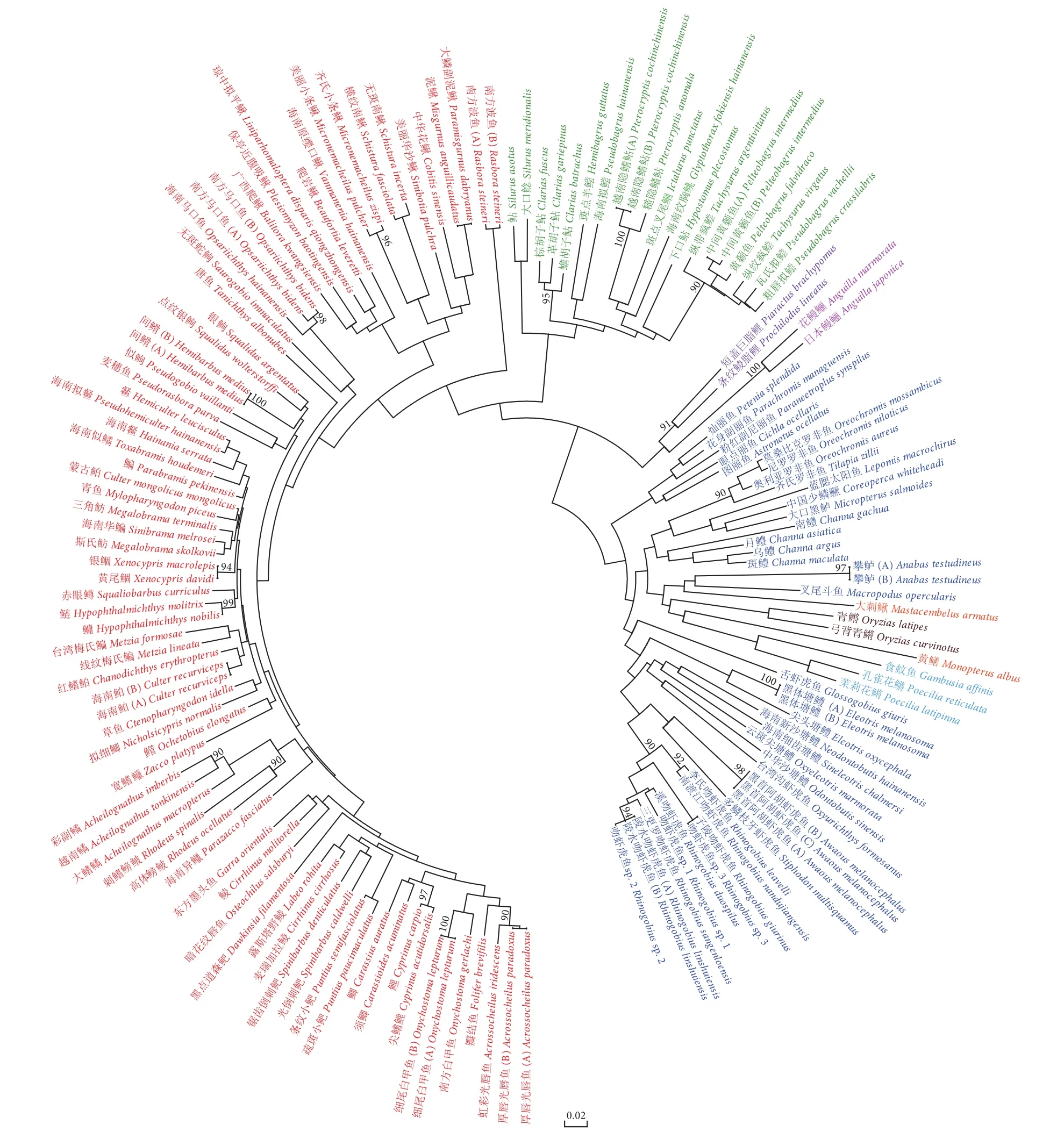
图5 基于150条线粒体16S序列构建的NJ系统发育树Fig. 5 NJ phylogenetic tree constructed based on 150 mitochondrial 16S sequences
3 讨论
3.1 数据库的构建原则
本研究的结果表明,数据库的构建应遵循“自建为主、公共为辅,两者结合”的原则。海南岛地理位置相对孤立,生物种类及特有类群均比较丰富[19]。虽然自建数据库在物种覆盖度上低于公共数据库,但综合附录A及申志新等[20]和李高俊等[21]的研究可以发现:公共数据库缺失的物种主要是海南岛当下确有分布的土著特有种、少见种和地理亚种[21],而自建数据库对这部分鱼类却有较好的收集——72种鱼类中,土著特有种就多达11种。公共数据库内现存土著特有种参考序列的缺失 (如海南异鱲、海南华鳊等) ,对当前海南岛淡水鱼类多样性的调查极为不利。在本研究中,基于公共数据库有30.56%~33.33%的物种比对不到高相似度序列,这直接导致基于公共数据库的物种注释准确率明显偏低。Jerde等[28]和Lim等[29]的研究也表明,NCBI数据库对地方特有种的条形码收集十分不健全,难以满足土著淡水鱼类多样性的调查需求。Gillet等[30]从少见种鉴定角度出发,也认为只有先自建参考数据才能开展后续多样性调查。对于常见种,虽然公共数据库已收录了较多参考序列 (表1),但其中明确标注采样地点的序列总数较少,这降低了条形码序列的参考价值。并且,由于参考序列存在定种错误 (如同物异名) 等原因,导致基于公共数据库比对到的候选物种数量更多[28,31](比如本研究中的COI基因)。候选物种越多,则序列注释受到错误信息的干扰程度越大[28]。特别是当物种存在同域分布记录时,难以判定序列所代表的真实物种[16,28]。基于公共数据库的注释准确率明显低于自建数据库 (表3)。从上述角度考虑,实地采集样品自建数据库几乎是鱼类环境DNA宏条形码研究中不可替代的步骤。
然而,自建数据库的缺点也十分明显——建库过程费时费力。本研究72种鱼类的获取耗时约3年;而从公共数据获取数据耗时却不超过24 h。即使如此长期采样,本研究中的自建数据库仍仅覆盖了研究水域约一半的鱼类。Miya等[32]提及了2012—2020年20多篇基于环境DNA宏条形码技术的鱼类多样性研究,也未见有高通量测序OTUs能被自建数据库完全覆盖和/或注释的报道。由此可见全面、准确的参考数据库构建难度之大。从物种覆盖度角度考虑,公共数据库无疑是自建数据库的有效补充。
自建国以来,海南岛仅进行了2次全面系统的淡水鱼类多样性调查,记录的鱼类种数分别为106和124种[20]。本研究实地采集样品72种,公共数据库补充67种。这139种鱼类最大限度覆盖了海南岛的已知淡水鱼类种数。然而,仍有部分鱼类难以获得其条形码参考序列,如原田鳑鲏(Rhodeus haradai)、大鳞鲢 (Hypophthalmichthysharmandi)、戴氏虾虎鱼 (Rhinogobius davidi) 等 (附录A)。这部分鱼类,仅有历史记录,实际已多年未采集到样品[20-21],也未在公共数据库见到任何参考序列,很可能成为海南岛淡水鱼类环境DNA宏条形码的永久缺憾。
3.2 最优目标基因
本研究表明对海南岛淡水鱼类判别能力最高的目标基因为COI,其次为16S和12S。特别是以共有物种为注释对象、以自建数据库为本底资料时,COI基因注释准确率高达100%。以公共数据库为参考时,虽然COI序列比对过程中的候选物种最多 (图2),需要查阅更多的参考文献才能精准排除错误的候选物种,但该基因在公共数据库中物种覆盖度最高,参考序列最为丰富,实际注释准确率也仅次于16S (表3)。这一结果与标准的脊椎动物DNA条形码普遍采用COI基因的现实相符合[33]。
然而,鱼类环境DNA宏条形码研究却很少以COI为目标基因[16]。其中最大问题是COI基因很难设计出短片段通用性引物[17]。Balasingham等[5]专门针对欧洲鱼类设计了COI通用引物——PS1,该引物对北京水体的鱼类检出数为MiFish-U的82.26% (51/62)。序列比对结果表明,PS1正、反向引物分别与海南岛淡水鱼类序列存在3~4和4~6个错配碱基 (结果未展示)。本研究使用的COI条形码短片段引物,是基于148种鱼类的COI全序列,在改进PS1、Uni-Minibar和标准的COI条形码通用引物基础上专门针对海南岛淡水鱼类设计的。分子实验过程中,全部鱼类的PCR产物条带清晰、明亮;而PS1引物则有26种鱼类不适用(结果未展示)。本研究的建库案例表明在不过分要求通用性的研究中,以COI作为目标基因可以设计出环境DNA宏条形码通用引物。
Zhang等[16]全面比较了23对鱼类环境DNA宏条形码通用引物,发现从引物目标基因来看,12S优于16S。本研究得出的结果也与上述主流观点不同。这是因为以往的引物比较研究,评价指标主要为环境DNA宏条形码技术的鱼类检出数[16,34-35]。而本研究则侧重基于本底资料比较物种序列注释的准确性。MiFish-U为Miya等[17]基于880种海洋鱼类线粒体全序列筛选出的环境DNA宏条形码引物 ,其通用性自然高于COI 和Vert-16S引物[32](注:MiFish的通用性可能过高,实际应用过程中极易产生微生物、鸟类、哺乳类的非特异扩增)。但MiFish-U的扩增子片段长度约170 bp,低于 Vert-16S 片段的 256 bp。Balasingham 等[5]、Gantner等[36]的研究表明,12S和16S扩增子片段越长,物种鉴定的准确性越高。本研究结果 (表3)与上述结论相符合,16S对海南岛淡水鱼类的判别能力高于12S。
然而,Vert-16S引物也有一些通用性问题需要解决。在Zhang等[16]的研究中,Vert-16S 的鱼类检出数与PS1相同。本研究通过比较发现,Vert-16S引物主要针对非鱼类脊椎动物[24]。对于鱼类类群而言,其正向引物 (总长 24 bp) 序列的 15~17 bp处存在3个完全错配的碱基。引物近3'端存在错配碱基会严重影响扩增效果[37]。因此后续也需要对Vert-16S的正向引物进行改进,以提高Vert-16S的鱼类检出数。基于PS1和Vert-16S引物均未完全匹配海南岛淡水鱼类序列的事实,本研究认为COI和16S可以作为某个特定区域鱼类环境DNA宏条形码研究的目标基因,但需要对通用引物进行优化改进。
3.3 种间差异阈值
标准的鱼类COI条形码长度约650 bp[25-26,38],种间差异阈值为0.02 (2%)[33]。本研究所确定的COI基因种间差异阈值却仅为0.006 9。这是因为COI基因不同区域的变异速率也并不相同。目前基于COI基因设计的鱼类环境DNA宏条形码通用引物很少,且无一例外均位于标准的鱼类COI条形码近 5'端前 350 bp 范围内[7,15-16]。Collins 等[39]、Menning 等[40]、Jennings等[41]、Sultana 等[42]的序列比对结果均表明,COI基因只有在这一区域序列较为保守,适合设计短片段引物,其余区域变异速率过快。本研究所用的短片段引物同样位于这一区域,因此其种间差异较小,阈值远小于0.02。
COI基因采用0.006 9种间差异阈值能够对本底资料库中94.96% (132种/139种) 的鱼类进行准确判定。对比其他环境DNA宏条形码研究,这一数值已经非常高。本研究中所用的COI短片段引物扩增子长度为139 bp,仅能够容许种内不同个体间出现1 bp的碱基变异;而12S和16S的扩增子长度和总平均遗传距离均明显高于COI,理论上可容许种内个体存在1.5~2 bp的碱基变异[43]。然而,12S和16S的物种判别准确性却未高于COI。以139种鱼类为本底资料,12S基因的种间差异阈值为0.005 6,在3种目标基因中阈值最小,种内个体判定的容错率也最低 (表4)。造成这种现象的主要原因是12S、16S序列种间遗传距离为0的物种数是COI的2~3倍。Miya等[17]和Bylemans等[44]的研究表明:MiFish-U引物扩增子位于茎环结构 (Stem-loop structure) 的高变环区,变异速率却又因种而异。陈治[45]研究发现:MiFish-U扩增子片段呈现高度保守与高度变异并存的特点,约有1/3浙江近海鱼类 (46种/147种)的变异速率高于COI,甚至接近控制区 (D-loop)。Milan等[18]比较MiFish-U及自行设计的NeoFish_3引物对67种淡水热带鱼类的判别能力,认为MiFish-U扩增子变异不稳定,甚至直接未对MiFish-U提出种间差异阈值。当研究区域和类群不同时,12S种间差异阈值可能会有所变动。这种种间差异阈值的不稳定性,可能是影响MiFish-U引物广泛应用的一个重要负面因素。
Milan等[18]基于NJ系统发育树确定的NeoFish_3最佳种间差异阈值为0.55%,物种判定准确率为91.04% (61种/67种)。本研究3种条形码阈值及判别准确率与之接近。这表明受扩增子长度的限制,环境DNA宏条形码的种间差异阈值可能都比较小,在1%以下;同时受制于鱼类自身的遗传特性,该技术从源头上就难以对鱼类进行100%区分[14,17,43]。不应过分夸大种间差异阈值及环境DNA宏条形码技术的物种判别能力。
4 结论
本研究结论如下:1) 自建数据库在序列注释准确性上显著高于公共数据库,但仍需要以公共数据库为补充;2) COI、16S的物种判别能力高于12S;3) 建议使用 0.006 9 (COI) 、0.007 5 (16S) 和0.005 6 (12S) 作为海南岛淡水鱼类环境 DNA 宏条形码研究的种间差异阈值。
