一个X连锁隐性遗传的Joubert综合征家系
2022-06-18陆超罗冠君沈玥谭媛程婷婷曹宗富罗敏娜马旭赵勇
陆超,罗冠君,沈玥,谭媛,程婷婷,曹宗富,罗敏娜,马旭*,赵勇*
(1.国家卫生健康委科学技术研究所,北京 100081;2.国家人类遗传资源中心,北京 102206;3.广州中医药大学附属南海妇产儿童医院,佛山 528200)
Joubert综合征(OMIM#213300,Joubert syndrome,简称JBTS)是一种罕见的神经系统发育疾病,通常表现为常染色体隐性遗传,只有OFD1基因变异导致的JBTS为X连锁隐性遗传。1969年首次由Joubert Marie及其同事发现并报道[1]。JBTS主要的影像学特征为中脑呈“磨牙征”[2],主要的临床表现有小脑蚓部发育不良或缺如、发育迟缓、肌张力低、新生儿期的阵发性呼吸过度、呼吸暂停和/或眼球运动障碍,部分患儿中有视网膜发育不良、多囊肾、多指(趾)症和肝纤维囊肿等伴发症[3]。目前,已经发现至少40个纤毛相关基因与JBTS有关[4-5],而国内关于OFD1基因变异引起的JBTS报道较少[6-8]。2017年,广州中医药大学附属南海妇产儿童医院康复科收治的一例患儿表现出典型的小脑蚓部发育不全、张力减退、发育迟缓等症状,临床确诊为Joubert综合征。经全外显子组测序(WES)分析以及Sanger测序验证,最终确认其致病位点为OFD1基因上的c.2626delC。现将该患儿的临床特点及遗传学分析结果报道如下。
材料和方法
一、实验对象
患者来自广州中医药大学附属南海妇产儿童医院康复科,在知情同意的前提下采集了患儿及父母、亲属的外周静脉血各2 ml,并获取了先证者的临床资料。本研究经国家卫生健康委科学技术研究所伦理委员会批准(批准文号为201806)。
二、全外显子组测序及数据分析
用QIAamp DNA Blood MiNi Kit(Qiagen,德国)提取全血基因组DNA,取0.5 μg先证者的DNA,根据标准操作步骤,用SureSelect Human All Exon V6试剂盒(Agilent,美国)进行外显子组文库制备,并利用Novaseq 6000平台(Illumina,美国)进行2×150的双末端测序,下机后数据的比对、注释、筛选等分析过程见参考文献[9-10]。
三、一代测序验证
依据候选致病性变异位点的物理位置,用NCBI blast在线工具(https://www.ncbi.nlm.nih.gov/tools/primer-blast)设计引物,由北京六合华大基因科技有限公司合成引物。引物序列分别为:正向引物5’-TGGTGAGGGTGACGTATTTG-3’,反向引物5’-ACAAGGAGAGGACAGGGATG-3’。以先证者及其家属的基因组DNA为模板,进行聚合酶链式反应,退火温度为65℃,随后用3730xl测序仪(ThermoFisher,美国)进行Sanger测序,用Chromas软件查看测序峰图,用SeqMan软件比对测序结果。
结 果
一、患者资料
1.基本情况:患儿,男,2岁,因“发现运动、智力发育落后1年余”由家长抱送入院行康复治疗。患儿为母亲的第4胎第3产,足月剖宫产,出生体重4.45 kg,无新生儿窒息、黄疸史。患儿1岁时表现出运动及智力发育迟缓,肢体松软。家系情况:父母非近亲婚配,先证者母亲曾自然流产一男胎,先证者外婆曾夭折一名男婴,原因未明,先证者兄姐均为正常表型。家系图谱详见图1。
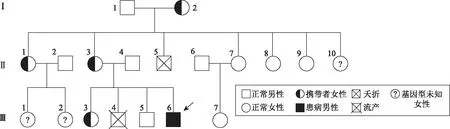
箭头所指为先证者图1 患儿家系图
2.体格检查:双手手指短,手掌肥厚,通贯掌纹;头控稳,能完成翻身、独坐、四爬,仅能扶物站立及行走;双手可主动抓物,对指捏物完成较笨拙;有一定目光交流,但反应迟钝,仅能指认部分常见物品,未能指认家人,仅能理解极个别简单指令,基本无语言表达能力;四肢肌力及肌张力低下,膝腱反射可引出,病理反射未引出。
3.辅助检查:颅脑MRI显示第四脑室变形,双侧大脑半球皮层下白质内及双侧侧脑室旁示多发斑片状等T1长T2异常信号影,脑白质髓鞘化进程中。小脑上脚增粗、延长,呈“磨牙征”(图2A),小脑蚓部形态失常。Gesell发育量表检查结果:社会适应DQ=47.4、大动作DQ=23.4、精细动作DQ=39.6、语言DQ=31、个人社交DQ=24.1、SM=8分。
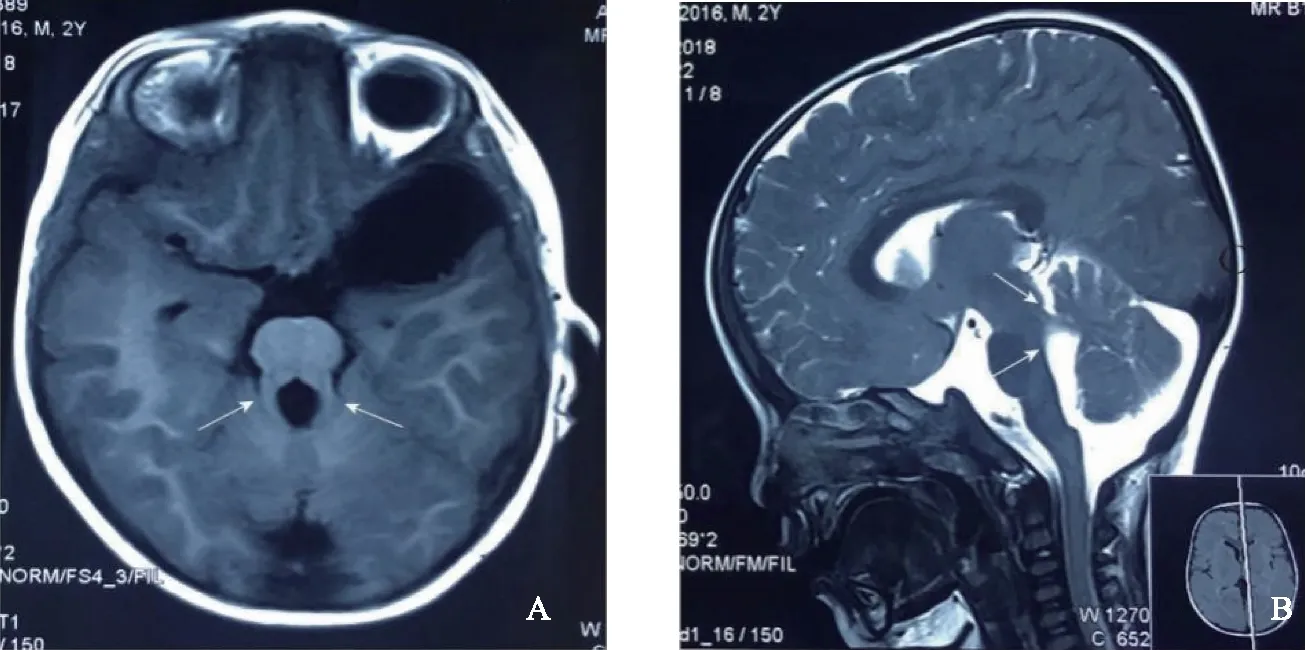
A:轴位T1WI显示“磨牙征”(箭头指示处);B:矢位T2WI显示小脑上脚增粗、延长(箭头指示处)。图2 患儿脑部磁共振成像(MRI)图
二、全外显子组测序及变异位点分析结果
原始数据经过比对、注释、过滤、筛选后,在已知致病基因中发现了OFD1基因(NM_003611.2)的c.2626delC变异,该变异位于第20号外显子,该碱基缺失导致第876位的谷氨酰胺变为赖氨酸,并造成蛋白质翻译提前终止(p.Gln876Lysfs*12),形成截短的蛋白质。该位点在dbSNP和HGMD均未收录,是尚未报道的新变异。根据ACMG解读指南,该位点为致病性变异。
三、Sanger测序验证
Sanger测序结果表明,先证者OFD1基因上的c.2626delC位点来自母亲(图3箭头所示)。父亲(Ⅱ4)为野生型半合子,母亲(Ⅱ5)为杂合子,先证者(Ⅲ6)为突变型半合子,先证者之姐为杂合子,先证者之兄为野生型半合子,符合伴X染色体隐性遗传定律。由于伴X染色体隐性遗传病的特殊性,后期回溯病人家族史,发现该家系中先证者母亲有姐妹5人(图1)。应亲属要求,对该家系育龄女性(Ⅰ2,Ⅱ1,Ⅱ7,Ⅱ8,Ⅱ9)进行了一代测序筛查,发现其家族中外婆(Ⅰ2)和大姨(Ⅱ1)均为该突变的携带者(图3)。
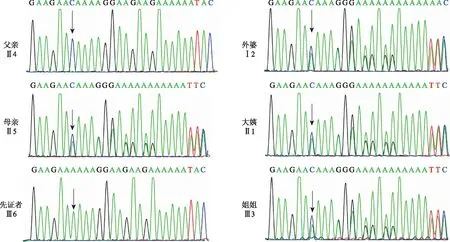
黑色箭头所示为变异位点所在的碱基图3 OFD1基因c. 2626delC位点的Sanger测序峰图
讨 论
OFD1基因变异被证实与多种疾病相关,包括口面指综合征Ⅰ型(OFD1,OMIM#311200)、Simpson-Golabi-Behmel综合征2型(SGBS2,OMIM#300209)、视网膜色素变性23型(RP23,OMIM#300424)和Joubert综合征[11-14]。OFD1最早被发现引起口面指综合征Ⅰ型[11]。口面指综合征Ⅰ型是一种X连锁显性遗传病,男性致死,女性患者通常有口腔畸形(上唇正中裂、腭裂、分叶舌、口腔系带增生、舌错构瘤、牙槽嵴增厚及牙列异常等)、特殊面容(前额突出、塌鼻梁、眼距宽等)和多指(趾)畸形等临床表现,中枢神经系统异常和多囊肾也是该病常见伴发症[15-18]。OFD1导致的SGBS2为X连锁隐形遗传病,患者表现为精神发育迟滞、肥胖、反复呼吸问题以及大头畸形等[12,19]。2009年,Coene等[14]首次报道OFD1与JBTS有关,该病患者均为男性,有“磨牙征”、发育迟缓、多指、视网膜色素变性等表型。这些因OFD1基因变异导致的疾病表型复杂多样,不仅在不同疾病类型的患者之间具有差异性,在相同疾病的患者之间,甚至在携带相同致病变异的患者中,其临床表现也可能存在差异[20]。本文患儿男性,头颅MRI呈现“磨牙征”、发育落后,全外显子组测序以及Sanger测序结果表明OFD1 c.2626delC是患儿的致病原因,为一例典型的X连锁隐性遗传JBTS病例。
OFD1基因位于X染色体短臂p22.2,包含23个外显子,编码一个包含1 012个氨基酸的蛋白质[21-22]。OFD1蛋白的主要功能区包括LisH结构域(72~101氨基酸,位于3号外显子)、7个coiled-coil结构域(位于7~21号外显子)以及5个无序结构域(IDRs)(719~991氨基酸,位于16~23号外显子),其中N端的LisH结构域在所有物种中高度保守[11,23]。目前,HGMD数据库中收录的OFD1基因的致病性变异中,与口面指综合征Ⅰ型相关变异138个,均位于17号外显子以前的位置。JBTS相关变异16个中8个分布在10号外显子以前,其余均在20号外显子以后的位置,本文的致病变异也在20号以后的位置上。有文献推测OFD117号外显子前发生的无义突变,尤其是发生在10号外显子前的无义和错义变异可能导致了OFD1蛋白质功能失调,而发生在OFD117号外显子后的变异造成了疾病类型的多样(如SGBS2和JBTS)以及疾病表型的多变和温和[23-26],这可能是本文患儿表型较为轻微的原因之一。2017年,孟晨等[6]曾报道一例因OFD1变异的男性Joubert综合征患儿,其变异位点为c.2843_2844delAA,位于OFD1基因21号外显子上。该患儿的临床症状除中脑呈“磨牙状”外,还伴有多指、四肢短小、视盘缺损、内脏反位的临床症状。2020年,Zhang等[8]发现一个因OFD1基因7号外显子c.599T>C变异所致的JBTS家系,该变异靠近OFD1基因N端的位置。先证者具有“磨牙征”,主要表现为重度发育迟缓和智力障碍。其母再孕2次均为患胎,基因型与先证者同,且第4胎除“磨牙征”外还有法洛四联症的影像学表现。本文患儿的致病变异发生于20号外显子,虽同样是靠近OFD1基因C端的位置,但较孟晨等[6]报道的病例症状为轻,从另一方面证实了该基因所致疾病表型的差异性。
本文报道的先证者目前暂未发现其他伴发症状,需要关注的主要是发育迟缓,干预手段主要是康复治疗。后期询问家族史时,我们发现先证者母亲有多位女性同胞,考虑到OFD1伴X隐性遗传的特征,女性携带者生育时,男婴的患病风险高达50%,女婴则有50%的可能为携带者,因此对该家族女性进行OFD1 c.2626delC变异位点的携带者筛查非常必要。告知筛查意义后我们应亲属要求为该家系育龄女性进行了基因筛查,发现先证者大姨为c.2626delC变异的携带者(图1)。先证者大姨的两个女儿因未成年暂未接受携带者检测,待其婚配时,可再行检测以确定是否携带c.2626delC变异。一旦确定携带OFD1致病位点,则孕期需进行产前诊断,以避免JBTS患儿的出生。因此,本文研究结果对于该家系有生育需求的女性携带者的孕前遗传咨询和产前诊断具有重要意义。
综上,本文利用全外显子组测序技术发现了一个新的OFD1基因的致病变异,扩大了OFD1基因的突变谱。本文是由OFD1基因变异引起的多成员JBTS家系,基因检测结果可为该家族女性携带者的优生优育提供重要的分子依据。
