吲哚化合物C-2位烷基化反应研究进展
2022-06-15范明会邹良邦田凯迪
郦 勇 范明会 邹良邦 田凯迪 程 凯
(1. 浙江省精细化学品传统工艺替代技术研究实验室,浙江 绍兴 312000;2.绍兴文理学院 化学化工学院,浙江 绍兴 312000)
0 引言
吲哚结构因其与众多受体的优秀结合能力,作为药物设计中的“优势骨架”(privileged scaffold)广泛存在于抗肿瘤、抗疟疾、抗病毒等药物分子及前体中[1].同时,吲哚也是天然产物分子中极其常见的结构单元(见图1).多年来,有机化学家投入了巨大的精力用以发展吲哚官能团化的方法.这其中,针对吲哚C-2位的官能团化方法的研究相对较少,而该位置直接烷基化反应的研究报道则更加有限.近十年以来,基于C—H键活化的方式来实现吲哚C-2位官能团化的研究有了显著进展[2-3].其中,多种新型的过渡金属催化剂(Pd、Ru、Ni等)被开发用以实现C—H键活化之后的烷基化反应.另外,自由基取代反应也是实现吲哚C-2位烷基化的传统手段[4-5],而近期多种光介导的新型自由基反应体系也被应用于合成吲哚C-2位的烷基化产物.此外,吲哚C-2位的碳负离子也可以在硼试剂的促进下发生相应的烷基化反应.本文总结了2013年以来新型的吲哚C-2位烷基化方法,并依照其反应类型进行综述介绍.
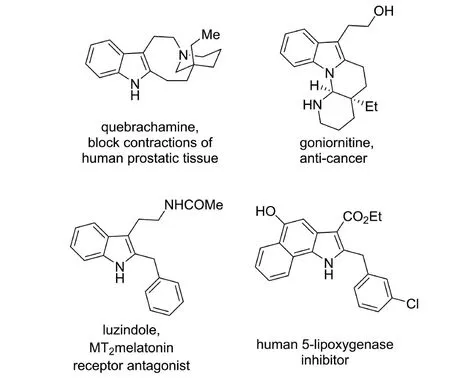
图1 几个具有生物活性的吲哚化合物
1 基于C—H键活化的吲哚C-2位烷基化
近10年来,有机化学家发展了多种基于C—H键活化的吲哚C-2位烷基化方法学.这些转化方法一般以吲哚N-1或C-3位上的官能团作为导向基团,在Pd、Ru、Ni、Ir、Mn这些过渡金属配合物的作用下,先进行C-2位的C—H键活化,然后与烷基卤化物发生氧化加成,再通过还原消除生成C-2位的烷基化产物.
2014年,Bach课题组[6]报道了Pd/降冰片烯共催化的1H吲哚化合物C-2位选择性烷基化反应(图2).该反应以1H-吲哚化合物1为底物,伯烷基卤化物2为亲电试剂,PdCl2(MeCN)2和降冰片烯作为共催化剂,在60 ℃~90 ℃下以良好的产率(46%~90%)得到2-烷基-1H-吲哚化合物3.反应过程中吲哚N-1与钯、降冰片烯相互作用,从而选择性地活化了吲哚C-2位的C—H键,生成了钯杂环中间体A.该反应底物适用性较广,可以在吲哚C-2位引入含有保护的羟基、环丙烷、酯基、缩醛、氰基等官能团的伯烷基侧链,同时可以容忍吲哚C-5或C-6有卤素、硝基、酯基等多种基团.对于吲哚C-3位有简单烷基侧链的底物亦可以顺利反应.
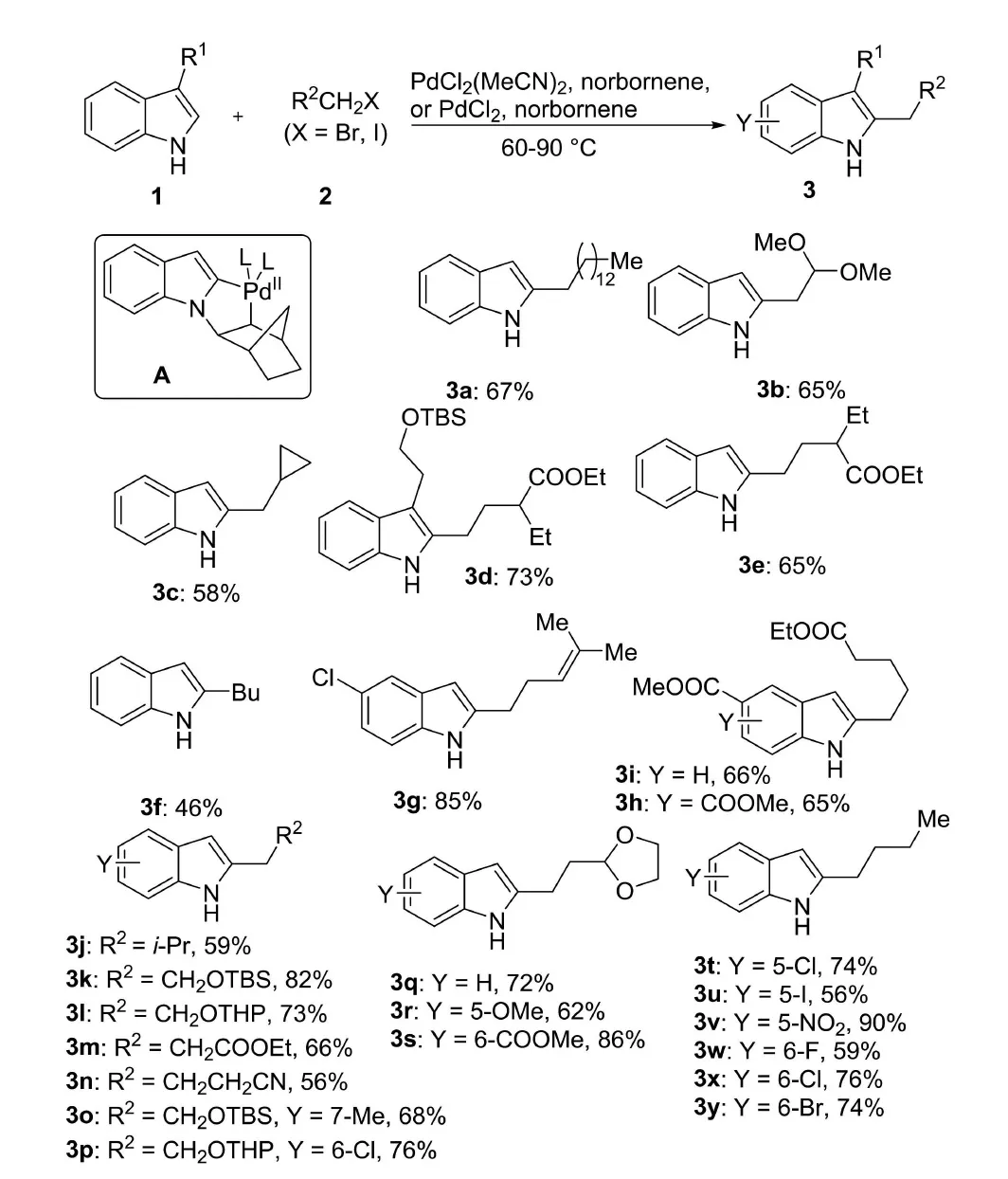
图2 1H-吲哚与伯烷基溴的烷基化反应
同时期,Bach课题组[7]还报道了Pd(II)/降冰片烯介导的N-叔丁基氧羰基(Boc)保护的(S)色氨酸乙酯吲哚的C-2位的区域选择性烷基化反应(见图3).该反应在60 ℃下,以Boc保护的(S)色氨酸乙酯吲哚化合物4为底物,可以在吲哚C-2位上引入一系列含有保护的羟基、环丙烷、苯基、缩醛、氰基等官能团的伯烷基侧链,且反应在氨基酸的立体中心不发生外消旋作用.值得注意的是,该反应条件不适用于仲或者叔烷基侧链的引入.
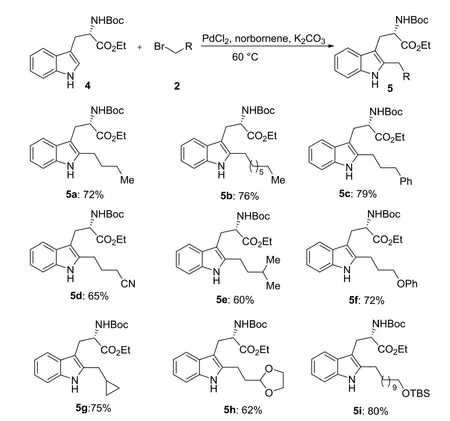
图3 色氨酸酯与伯烷基溴的烷基化反应
2015年,Prabhu课题组[8]使用吲哚N-1上的苯甲酰基(Bz)作为导向基团,在Ru(II)催化体系下对吲哚2位进行C—H键活化,进而实现了吲哚C-2位的选择性官能化,并引入了丁二酰亚胺和丁二酸酯片段(图4).在该反应中通过选择合适的烯烃化合物(马来酰亚胺和马来酸酯)来避免β-H的消除反应,从而得到了共轭加成产物,而非Heck型产物. 反应以[Ru(p-cymene)Cl2]2/AgSbF6/Cu(OAc)2·H2O为催化体系,醋酸作为添加剂,二氯乙烷(DCE)为溶剂,反应温度为120 ℃,以50%~81%的产率合成了C-2丁二酰亚胺或丁二酸酯取代的吲哚化合物8和10.

图4 钌催化的吲哚C-2位烷基化反应
2016年,Punji[9]报道了一种镍催化的单齿螯合吲哚直接C-2烷基化反应(图5).在(喹啉基)氨基镍配合物[κNκN,κN-{Et2NCH2C(O)-(μ-N)-C9H6N}Ni(OAc)]14的催化下,催化量的双三甲基硅基胺基锂(LiHMDS)和当量的叔丁醇锂(LiOtBu)作用下,N-2-吡啶(py)或N-2-嘧啶(pym)吲哚化合物11与卤代物12在甲苯中发生偶联反应,从而以38%~93%的产率在吲哚C-2引入伯、仲烷基侧链,得到产物13.该方法具有充足的底物范围,可容忍吲哚上的卤素、烷基、甲氧基、氰基等基团的存在.

图5 镍催化的吲哚化合物的C-2位烷基化反应
2017年,Punji课题组[10]进一步发展了在(喹啉基)氨基镍催化剂[κNκN,κN-{Et2NCH2C(O)-(μ-N)-C9H6N}Ni(OAc)]14的催化下,氧化剂2-碘丁烷的作用下,甲苯衍生物与吲哚化合物在C-2位的氧化偶联反应(图6).该反应中,2-碘丁烷可通过从甲苯衍生物中提取H原子,从而起到苄基起始剂的作用.反应可以高效合成2-苄基吲哚化合物16,产率最高可达98%.反应底物适用性较为广泛,可以容忍吲哚C-2位带有多种取代基的苄基,如卤素、甲氧基、烷基等基团;亦可容忍吲哚底物中C-3位的甲基及C-5位的甲基、卤素等基团的存在.机理研究表明,反应中的苄基化过程通过苄基自由基的单电子转移来实现.
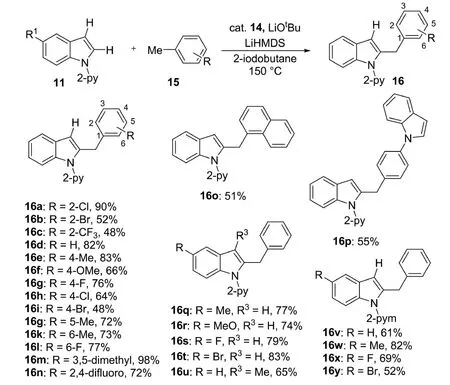
图6 镍催化的甲苯衍生物与吲哚化合物的C-2位苄基化反应
2020年,Fang课题组[11]利用吲哚上的N-2-吡啶作为导向基团,在Rh(III)配合物的催化下,选择性地在吲哚C-2位引入丁二酰亚胺及其衍生物片段(图7).该反应在120 ℃下进行,以41%~96%的产率得到了N-2-吡啶吲哚烷基化产物17.底物适用性广泛,可以容忍吲哚上卤素、酯基、甲氧基、氰基、硝基等官能团,亦可引入带有苯基、酚羟基、烷基等基团的丁二酰亚胺以及马来酸酯、丙烯酸乙酯等衍生物片段.值得注意的是,该反应只需0.5 mol%~1.0 mol%催化剂[RhCp*Cl2]2,即可在无添加剂的条件下顺利进行.

图7 铑催化的吲哚化合物与丁二酰亚胺及衍生物的烷基化反应
2015年,Shibata课题组[12]发展了以吲哚C-3位芳酰基作为导向基团,在手性二膦配体(SEGPHOS或xylylBINAP)和阳离子铱催化剂的作用下,实现了N-烯基吲哚的分子内对映选择性烷基化反应(图8).在吲哚底物C-3位侧链上芳酰基的导向作用下,手性Ir配合物活化C-2位的C—H键,而后与吲哚N侧链上的烯烃发生加成环化.在120 ℃~135 ℃下,反应以良好到优秀的收率(61%~98%)和优秀的对映体过量值(ee,最高可达98%)生成1-取代-2,3-二氢-1H-吡咯[1,2-α]吲哚化合物19.除了端烯底物之外,含有Z式二取代烯烃的20、22同样可以在该反应下顺利环化,从而分别生成高对映选择性的产物21和23.

图8 铱催化N-烯基分子内对映选择性环化反应
2020年,Punji课题组[13]报道了一种在非格式试剂的条件下,Mn配合物介导的吲哚化合物C-2位选择性烷基化反应(图9).该方法使用简单廉价的MnBr2作为催化剂,2,2′-联吡啶(bpy)为配体,LiHMDS为碱,在90 ℃下可以实现N-2-py-吲哚化合物和碘化物在C-2位的偶联,以38%~91%的产率生成烷基化产物29.该方法适用于含有卤素、甲氧基等多种取代基的吲哚底物以及带有卤素、烯基、吡咯和咔唑基等多种官能团的碘化物,且二卤化物也可顺利发生反应.初步的机理研究表明,烷基化过程中C—I键的锰插入过程可能为单电子氧化加成反应.

图9 锰介导的吲哚C-2位烷基化反应
2 基于自由基的吲哚C-2位烷基化
吲哚化合物的烷基自由基取代反应通常发生在C-2位,其原因是烷基自由基加成在吲哚底物的C-2位后可以生成相对稳定的苄基自由基.因此,自由基类型的反应一直是实现吲哚C-2位烷基化的重要手段.
2014年,Melchiorre课题组[14]提出了一种无金属介导的光化学方法可以直接实现吲哚烷基化(图10).反应过程中,取代的1H-吲哚化合物(电子给体)与α-羰基或苄基溴化物(电子受体)结合生成了电子给体-受体配合物(EDA).在节能灯(CFL)的照射下,EDA发生单电子转移,生成了吲哚自由基和苄基自由基.两者相互结合得到了烷基化产物31,而反应产生的副产物氢溴酸则需要添加2,6-二甲基吡啶来中和.值得注意的是,只有C-3位有取代基的吲哚底物才能在C-2位发生烷基化反应.该方法可以容忍吲哚上的烷基、酯基、苯基等官能团,产率最高可达93%.

图10 无金属介导的光化学吲哚烷基化
2019年,Gryko课题组[15]介绍了吲哚化合物与重氮酯的光烷基化反应(图11).在蓝光照射下, 负载量仅为0.075 mol%的光催化剂Ru(bpy)3即可以催化吲哚底物32和重氮酯33发生反应,从而顺利生成吲哚C-2烷基化产物34.该反应条件适用于连有烷基、甲氧基等供电子基团的吲哚底物和连有酯基、磷酸酯等吸电子基的重氮酯.而对于连有N-Boc、氰基、溴等吸电子基的吲哚底物,则需要另加入N,N-二甲基-4-甲氧基苯胺(35)作为Ru配合物的共催化剂.该方法整体上适用于各类型的吲哚底物,产率最高可达到90%.
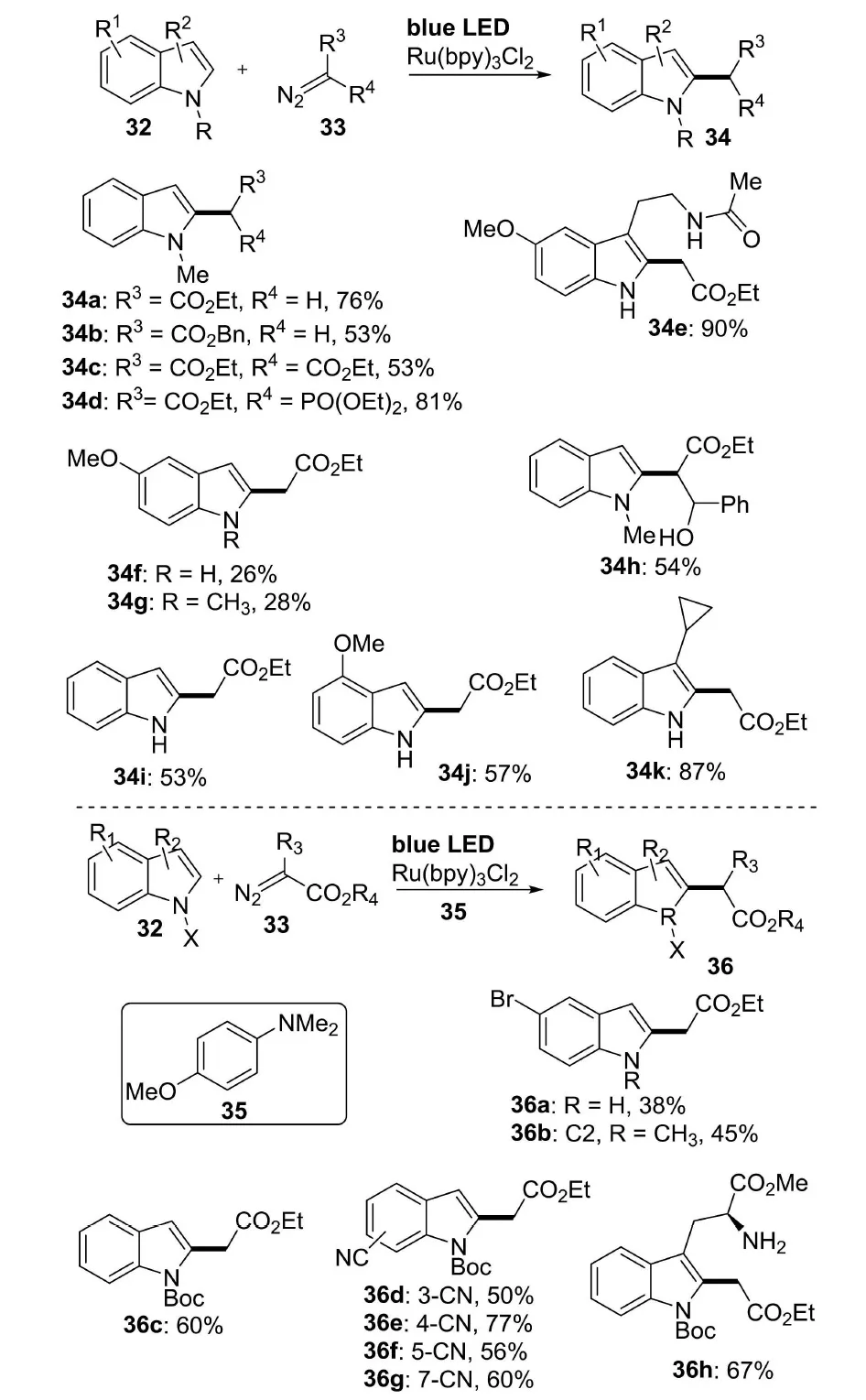
图11 吲哚化合物与重氮酯的光烷基化反应
2021年,Paixão等人[16]报道了在可见光照射下, 用活化的α-溴代羰基化合物对色氨酸和含色氨酸的肽进行高效选择性的C—H官能化反应,以合成多肽的烷基化产物.该反应在光照下,室温条件下以Ir{dFCF3ppy}2(bpy)PF6为催化剂,2,6-二甲基吡啶为碱,以21%~76%之间的产率得到了烷基化产物.反应条件对α-溴苯乙酮、酯类、硫酯类、酰胺类和氨基酸等烷基溴化物具有耐受性(图12).同时,该反应能够耐受包含亮氨酸(Leu)、缬氨酸(Val)、甘氨酸(Gly)、丙氨酸(Ala)和苯丙氨酸(Phe)残基等的色氨酸二肽、三肽和四肽,包括含有羟基或羧基的多肽.此外,通过将三肽底物与含有药物分子的工程含溴二肽连接,组装出高度功能化的多肽(药物肽偶联物),如连有阿尔茨海默病药物美金刚胺(Memantine)的39s,连有癫痫药物预加巴林(Pregabaline)的39t.
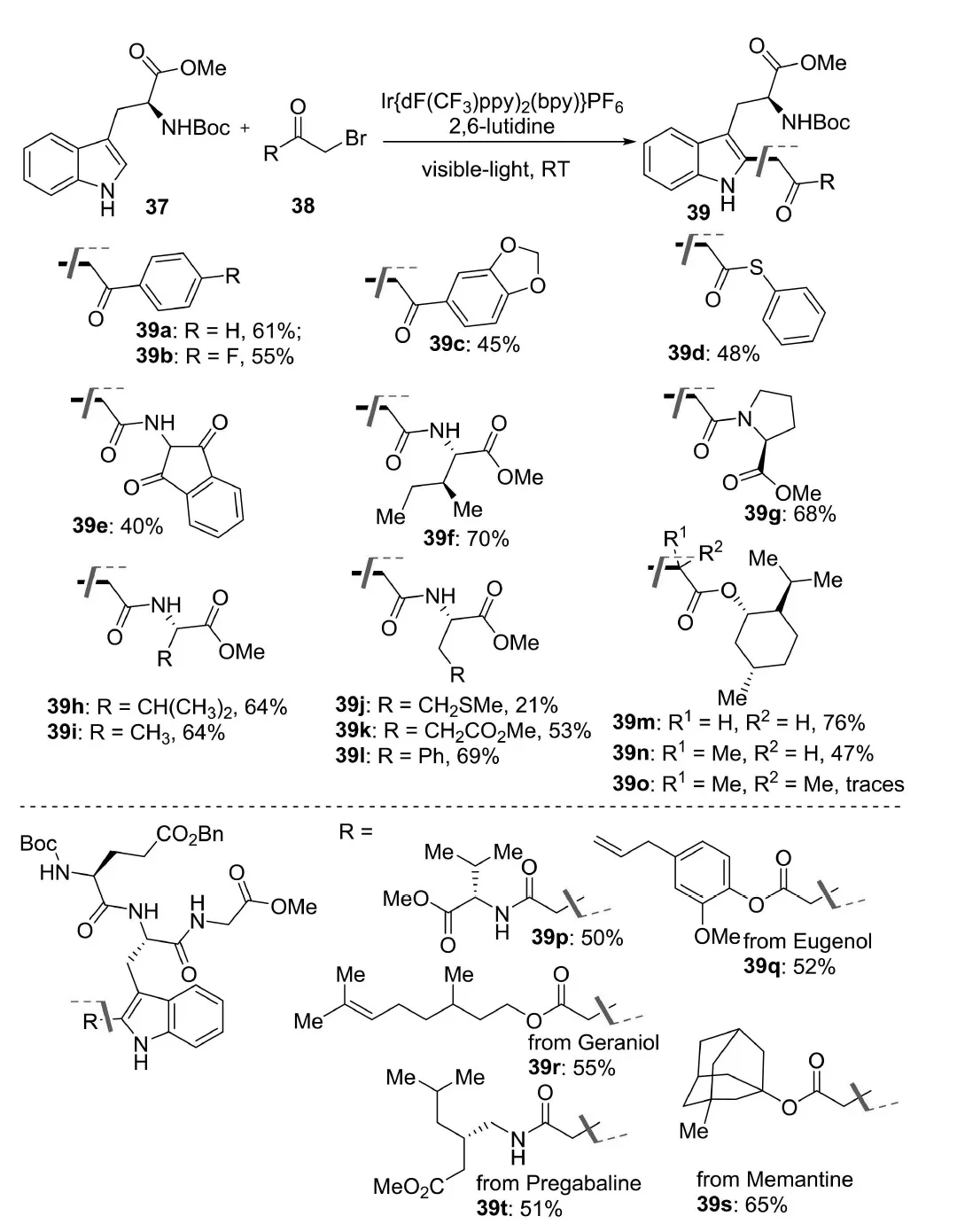
图12 光催化色氨酸衍生物的吲哚C-2位烷基化
3 硼试剂介导的吲哚C-2位烷基化
2018年,Aggarwal课题组[17]报道了吲哚基硼酸盐配合物41与碘代烷2在极性双分子亲核取代条件下反应(图13).在正丁基锂的作用下,吲哚底物和烷基硼酸反应得到了吲哚基硼酸盐配合物41.然后,41与烷基碘和单质碘反应生成了完全立体特异性的C-2和C-3位双烷基化产物.反应在室温下进行,产率最高可达82%.该反应底物适用性广泛,可耐受吲哚底物上的腈基、酮基、酯基、砜和酰胺基等官能团.初步的机理表明,富电子的吲哚硼酸盐具有较强的亲核能力,在极性条件下可以直接与烷基碘化物反应.
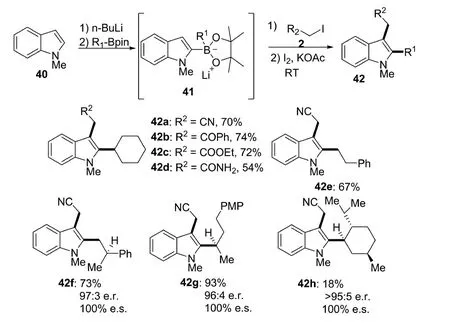
图13 硼试剂介导的吲哚C-2和C-3位的双烷基化反应
4 结论
吲哚作为药物化学中的重要结构单元,一直受到化学家和药物学家的广泛关注.同时,因其独特的双环结构,吲哚亦成为有机化学家探索新反应的优秀底物.文中吲哚C-2位烷基化方法学的发展丰富了吲哚衍生物的合成手段,提高了其合成效率.然而,以上方法学大都需要使用过渡金属配合物作为催化剂,烷基卤化物作为烷基化试剂.如今,随着环境保护、绿色化学等可持续发展理念日渐深入人心,使用更高效的合成策略,更绿色的催化体系和环境友好的试剂来合成吲哚衍生物必将成为未来有机化学家探索的重点方向.
