基于液相色谱-质谱联用技术的蛋白质类肿瘤标志物定量方法研究进展
2022-06-15樊小雪楚占营朱曼曼雷喜梅宋雨蒙武利庆戴新华俞晓平
樊小雪,翟 睿,楚占营,朱曼曼,雷喜梅,宋雨蒙,武利庆,江 游,戴新华,方 向,俞晓平
(1.中国计量大学生命科学学院,浙江省生物计量及检验检疫技术重点实验室,浙江 杭州 310018;2.中国计量科学研究院前沿计量科学中心,国家市场监管技术创新中心(质谱),北京 100029)
肿瘤标志物(TM)是一类可用于指示肿瘤发生、发展等过程的生物分子,主要存在于患者的体液(如血液、淋巴液等)和组织中[1],在肿瘤的早期筛查、良恶鉴别、预后评估及术后监测等方面发挥重要作用[2]。目前,肿瘤标志物的主要存在形式为蛋白质、核酸、多胺类等。蛋白质是生命活动的主要承担者,蛋白质类肿瘤标志物能提供诸多功能信息,并精准反映相关细胞的生理状态,因而成为研究肿瘤标志物的重要载体[3]。但蛋白质的丰度动态范围较宽,且存在复杂多样的翻译后修饰,为蛋白质类肿瘤标志物的准确定量提出了挑战[4-5]。
液相色谱-质谱(LC-MS)联用技术是鉴定和定量生物样品中蛋白质的有力工具,已逐步应用于许多医学相关的研究领域,如治疗药物监测、毒理学分析及蛋白质定量分析等[6]。基于LC-MS技术对蛋白质类肿瘤标志物定量的方法不依赖于抗体,并且能提供高度的特异性及异构体辨别能力,相较于基于抗体的方法(如免疫印迹法[7]、酶联免疫吸附法[8]等)有诸多优势,如可以高通量检测和量化天然蛋白质及翻译后修饰等[9]。如何在含有高丰度蛋白的复杂样品中准确检测和定量蛋白质类肿瘤标志物是基于LC-MS对蛋白质类肿瘤标志物定量的一个挑战[10]。研究人员通过不同的设计原理开发出多种定量方法,实现了高度复杂样品中蛋白质类肿瘤标志物的准确定量[11-12]。本文将从用于定量蛋白质类肿瘤标志物的样品前处理方法及质谱扫描策略2个方面,对近年来的主要定量方法进行总结。
1 用于定量蛋白质类肿瘤标志物的样品前处理方法
目前,基于LC-MS定量肿瘤标志物的前处理策略主要分为非标记策略和标记策略。根据引入同位素的方式,标记策略分为体内标记(如细胞培养条件下稳定同位素标记技术(SILAC))和体外标记(如等重同位素标记相对和绝对定量技术(iTRAQ)、串联质量标签(TMT)等)。
1.1 非标记策略
非标记策略(lable-free)是一种不需要使用稳定同位素标签的技术,通过对酶解后的肽段进行LC-MS/MS大规模数据分析,比较不同样品中相应肽段的信号强度,从而对肽段对应的蛋白质进行相对定量;也可以通过比较同一肽段质谱水平的离子强度或峰面积等进行定量[13-14]。与同位素标记方法相比,其具有灵活性和成本效益,且不涉及复杂的标记步骤等优势[15-16]。Yang等[17]采用非标记策略,通过鸟枪法定量分析实验组与对照组之间的糖蛋白表达差异,结果显示α-1抗胰蛋白酶是最具潜力的候选生物标志物之一。随后使用酶联免疫吸附试验测试了70个独立样本(35例膀胱癌病例),对候选生物标志物A1AT进行验证,实验表明,A1AT检测对膀胱癌的灵敏度为74%,特异性为80%。Song等[18]为了寻找对胃癌具有早期诊断价值的新型生物标志物,通过非标记策略进行了大规模蛋白质组学分析,示于图1,研究结果确定了537个差异表达的蛋白质(包括280种上调和257种下调蛋白质),筛选出15个潜在胃癌生物标志物,并提出4种蛋白质(ATP5B、ATP5O、NDUFB4、NDUFB8)对胃癌有较高的诊断能力,为胃癌的治疗提供了新靶点。
1.2 标记策略
1.2.1细胞培养条件下稳定同位素标记技术
细胞培养条件下稳定同位素标记技术(SILAC)定量的基本原理是采用稳定同位素标记的必需氨基酸取代细胞培养基中相应的氨基酸,细胞经5~6个细胞周期后,被同位素标记的氨基酸完全替代了原有氨基酸,参与细胞的蛋白质表达过程;将标记细胞与非标记细胞的裂解蛋白按细胞数或蛋白量等比例混合,经分离纯化后进行质谱鉴定,根据一级质谱图中2个同位素肽段的峰面积比进行相对定量[19]。本方法的优势是允许样品在细胞裂解消化成肽之前混合、标记效率高(可达100%)且标记效果稳定、灵敏度高、可直接实施定量,具有较高的准确性和可重复性。但SILAC技术是细胞层次的,对组织的定量难度较大且成本较高[20]。SILAC可用于分析恶性肿瘤的整体蛋白质组表达变化、阐明各种癌症恶性转化的潜在机制、研究药物或其他治疗引起的蛋白质组学变化等[21]。Lau等[22]通过对SILAC和稳定同位素二甲基标签法进行比较,发现SILAC更具重现性,且适用于大样本的蛋白质定量研究。Yeh等[23]采用SILAC和iTRAQ对蛋白质的表达水平进行分析,发现galectin-1可能是预测肝细胞癌(HCC)患者对索拉非尼治疗反应的生物标志物,并有望帮助HCC分期及指导个性化治疗。Clark等[24]通过三重SILAC定量分析策略,揭示了与非小细胞肺癌外泌体相关的蛋白表达谱,表明这些外泌体在肺癌发生发展中的作用,并确定了几种潜在的候选生物标志物,示于图2。
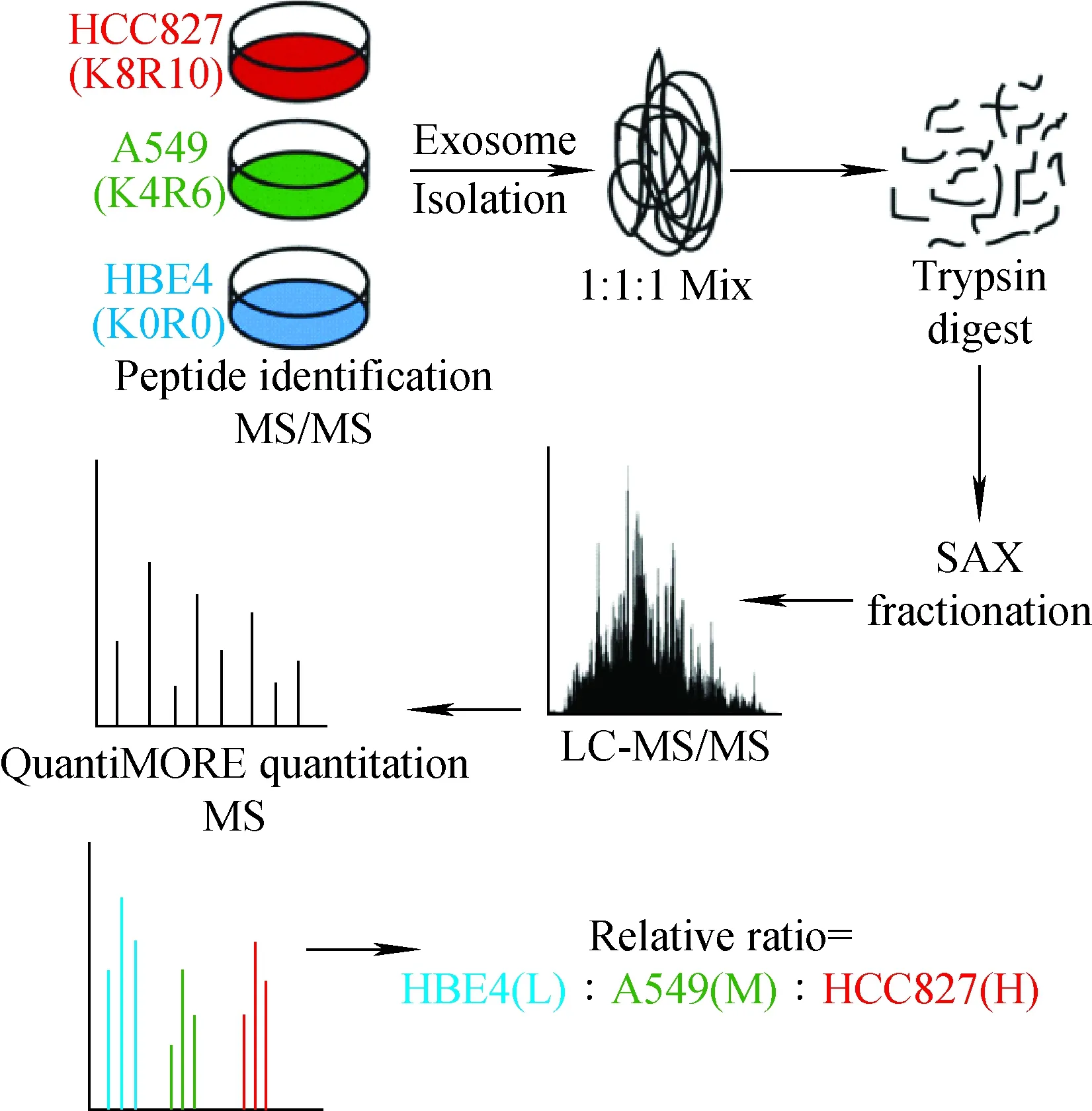
图2 三重SILAC定量分析策略示意图[24]Fig.2 Schematic diagram of triple SILAC quantitative[24]
1.2.2等重同位素标记相对和绝对定量 等重同位素标记相对和绝对定量(iTRAQ)[25]是基于iTRAQ标签的一种定量蛋白质组学技术,iTRAQ试剂是一种可与肽段氨基反应的等重同位素标签,包含报告基团、肽反应基团和平衡基团。一组iTRAQ试剂可同时标记4个或8个蛋白质样品,将标记的样品混合后进行LC-MS/MS分析[26-27]。在一级质谱中,不同样品中的相同肽段与相同质量数的iTRAQ试剂的反应产物具有相同的质荷比,表现为同一个峰。而在二级质谱中,iTRAQ标签的平衡基团断裂后,不同样品的报告基团表现出不同的质荷比,通过二级质谱中各报告基团峰的信号强弱实现肽段的相对定量,并回溯到蛋白质水平。iTRAQ技术具有很高的灵敏度和蛋白质覆盖率[28],可用于研究癌症的发生、发展以及复发/转移的分子机制[29-30],在肿瘤标志物的定量方面有多种应用,如追踪癌症术后早期复发的血清蛋白质组学[31]、分析定量癌症的候选生物标志物[32]等。Wang等[33]采用基于iTRAQ的定量蛋白质组学方法分析不同HCC亚型的分子特征,成功筛选出57种差异表达的蛋白质,它们在肿瘤组织和非肿瘤组织之间的表达程度显著不同,且与肿瘤大小密切相关。此外,Hsu等[34]使用基于iTRAQ标签的LC-MS/MS技术在1 763个蛋白质中鉴定并验证了6种潜在的肺腺癌生物标志物(ERO1L、PABPC4、RCC1、RPS25、NARS、TARS),这些蛋白质在无淋巴结转移的肺腺癌中的表达比相邻正常组织增加了1.5倍,且进一步研究发现,早期肺腺癌患者的ERO1L过表达与较差的生存率呈正相关。Xing等[35]基于iTRAQ的定量蛋白质组学方法系统地比较了单一和多发病变的原发性肝癌之间的总体蛋白质组概况,与非癌组织相比,多发病变和单一病变的HCC组织中分别有107和330个蛋白质表达异常;同时表明,两者的发生和发展可能存在不同的分子机制,HSD17B13和HK2蛋白可能是单一和多发病变的原发性肝癌的潜在生物标志物,示于图3。Xia等[36]采用iTRAQ结合二维液相色谱-串联质谱(2D LC-MS/MS)法鉴定出恶性间皮瘤患者和健康对照组间的145种差异蛋白,提出FLNA、FBLN1和TSP-1可能是诊断恶性间皮瘤及筛选高危人群的潜在蛋白质类候选标志物,并表明使用iTRAQ分析血清蛋白质组是发现生物标志物的可行策略。
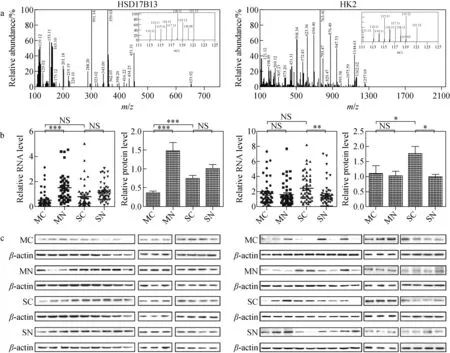
图3 基于iTRAQ的定量蛋白质组学方法系统地比较单一和多发病变的原发性肝癌之间的总体蛋白质组[35]Fig.3 Validation of the differentially expressed proteins in the primary HCC with single and multiple lesions[35]
1.2.3串联质量标签 与iTRAQ标签技术相似,串联质量标签(TMT)包括报告基团、肽反应基团、平衡基团3部分,反应基团与肽段的氨基发生特异性共价结合反应,将TMT试剂连接到肽段上。该技术最多可使用16个同位素标签来标记多肽的氨基。TMT技术具有高灵敏度、高通量等优点,可用于多种肽的分离[37-38]、肿瘤及其邻近组织中肽/蛋白质的定量[39]等。Huang等[40]使用10个同位素标签标记了6种乳腺肿瘤细胞裂解物中的蛋白质,并通过Q Exactive HF质谱对样品进行分析,共鉴定出8 706个蛋白质和28 186个磷酸肽,示于图4。2017年,Stewart等[41]使用TMT鉴定了5 535个蛋白质,表明TMT能够作为定量肿瘤标志物的方法。Dai等[42]在乳头状甲状腺癌(PTC)和侵入性表型(IPTC)上进行基于TMT标记的质谱蛋白质组学实验,鉴定并定量与PTC和IPTC组相同的4 607个蛋白质,发现了一些候选的生物标志物,为研究甲状腺癌的侵袭过程提供了新见解,为甲状腺癌的治疗提供了新思路。Zhou等[43]收集了15名早期胃癌患者和15名健康对照者的血浆样本,经选择性去除高丰度蛋白质后,用LC-MS/MS结合TMT对血浆样品进行分析,共鉴定出2 040个蛋白质,发现11个差异表达蛋白质,为早期胃癌的诊断提供了潜在的标志物,也为进一步探索胃癌的发病机制提供了线索。
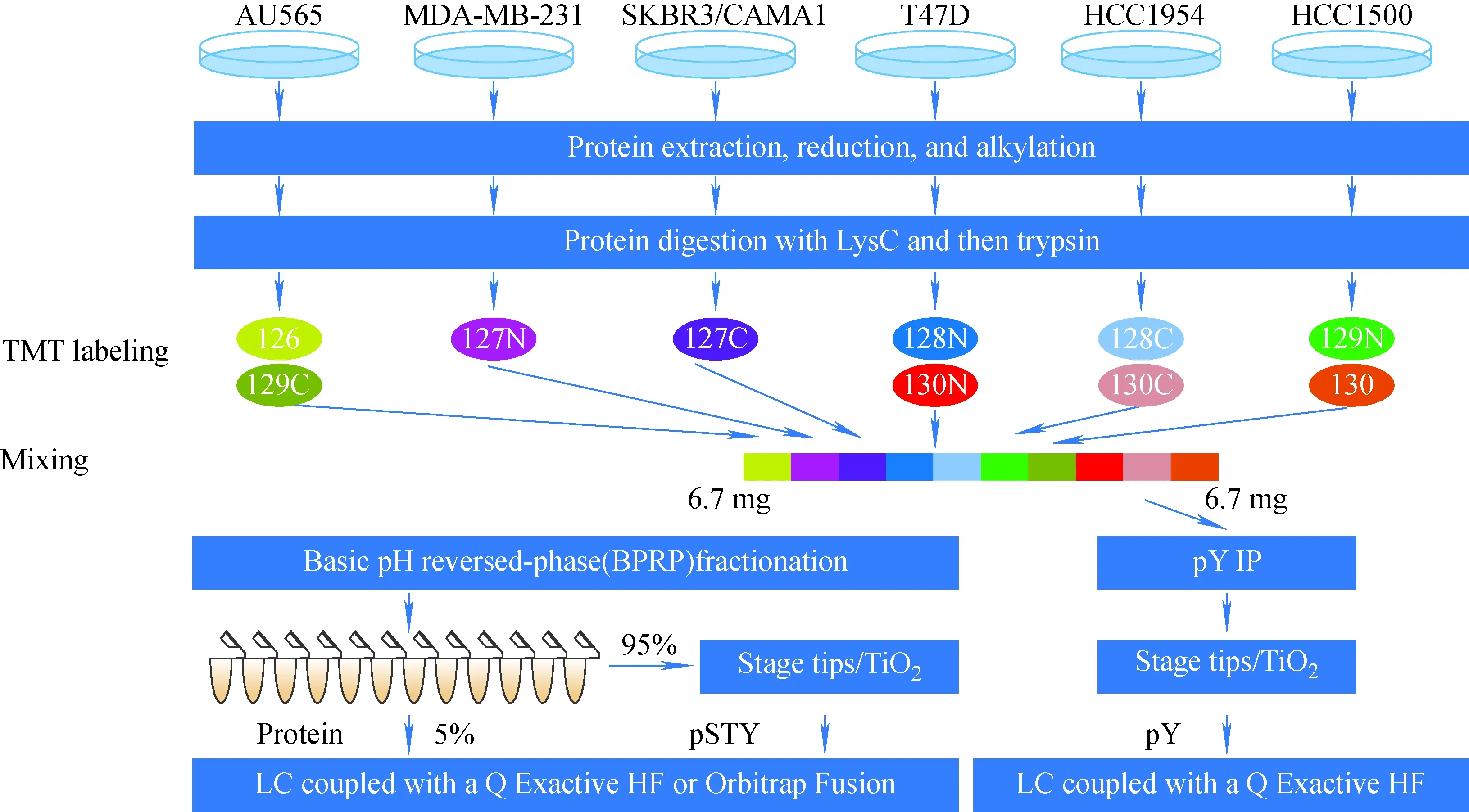
图4 酶切6种乳腺癌细胞裂解液中的蛋白提取物,酶切后的肽段用10种同位素标签标记,随后在质谱仪上分析样品[40]Fig.4 Protein extracts from the 6 types of breast cancer cell lysates were digested,and the digested peptides were labeled with 10 isotopic labels,and then the samples were analyzed on a mass spectrometer[40]
1.2.4稳定同位素编码亲和标签 稳定同位素编码亲和标签(ICAT)最早由Gygi及其同事提出[44],主要用于定量比较从同一生物系统的2个不同样本(状态)中提取的目标蛋白的相对表达水平。ICAT试剂由半胱氨酸反应基团、中间的连接子和亲和标签3部分组成,其分为轻标(不含氘)和重标(含8个氘)2种形式[45]。ICAT标签[46]首先选择性地标记含半胱氨酸的肽段,经LC-MS/MS分析,比较不同ICAT试剂标记的1对肽段的信号强度比例,定量计算其所对应蛋白质在细胞中的相对丰度。但ICAT无法检测缺乏半胱氨酸残基、强疏水性及翻译后修饰的肽,且用氘代ICAT试剂标记的多肽容易发生色谱峰分离,导致峰间比例不一致。Stewart等[47]使用ICAT比较顺铂敏感和耐药卵巢癌细胞系之间的差异蛋白组,发现63个蛋白质在顺铂敏感细胞中过表达,58个蛋白质在顺铂耐药细胞中过表达,表明一些蛋白质可能参与细胞对顺铂的反应,并可能作为卵巢癌的标记物及药物靶标。Kang等[48]采用ICAT和串联质谱法分析6例乳腺癌患者和6例健康女性的血浆蛋白组,共识别和定量了155个蛋白质,其中33个蛋白质在乳腺癌患者和健康女性的血浆之间表现出超过1.5倍的丰度变化,并通过实验将生物素酶确立为一种潜在的乳腺癌生物标志物。
2 LC-MS定量质谱扫描策略
蛋白质分子的相对分子质量通常高达数万甚至数十万,在质谱分析之前需进行酶解,最终从肽段的测量结果回溯到蛋白质含量。这一步骤使样品分析的复杂性剧增,一级质谱信息不足以对肽段进行定性,因而多级质谱扫描策略成为目前定量分析生物样品的有效方法。常用的定量质谱扫描策略主要包括多重反应监测(MRM)、平行反应监测(PRM)、数据独立采集(DIA)等[49],示于图5。

图5 MRM,PRM,DIA的工作原理[49]Fig.5 MRM,PRM,DIA working principle[49]
2.1 多重反应监测
多重反应监测(MRM)也称为选择性反应监测(SRM),通过对特异性母离子进行信号扫描,去除不符合要求的离子信号干扰,从而降低背景噪音、提高信噪比。MRM利用三重四极杆(QqQ)质谱优异的重复性、灵敏度和选择性,可有效区分蛋白质亚型、翻译后修饰及突变形式[50-51]。MRM具有临床及临床前研究所需的重现性高、定量精度高、生物分子分析速度快等优点[52-53],在分析目标肽段时通常需要使用已知质量的同位素标记肽段或蛋白质作为内标加入待测样本中,通过质谱仪测定同位素丰度比例,计算出目标蛋白质含量[54],并通过标准曲线对未知样品中的特定蛋白质进行绝对定量[55]。由于MRM仅能测量已知的蛋白质,不能做筛选性的分析,因此必须优化特定的操作条件[56]。目前,MRM技术已经参与了十几种血浆生物标志物的鉴定和验证[57]。Naboulsi等[58]采用非标记策略结合MRM方法分析了一个由50名患者组成的队列,证明了Versican核心蛋白(VCAN)与高分化低分期的HCC显著相关,首次证实了VCAN可作为早期HCC诊断的潜在生物标志物。Kim等[59]通过添加稳定同位素标记的蛋白质作为内标,使用单克隆抗体富集甲胎蛋白(AFP),使用具有高亲和力的LCA凝集素分离AFP-L3,之后进行去糖基化、胰蛋白酶消化及脱盐,比较MRM-MS与液相结合分析法(LiBA)的检测结果,证实采用MRM-MS方法检测AFP及AFP-L3的假阴性率较低,更适合肝癌的早期检测,示于图6。Kim等[60]应用LC-SRM对非小细胞肺癌的95种潜在肿瘤标志物进行分析,证实共有17个蛋白质可作为有效的肿瘤标记物,其中zyxin(ZYX)被确定为非小细胞肺癌的潜在早期诊断标志物。

图6 通过测定AFP和AFP-L3比较MRM-MS法和LiBA法[59]Fig.6 Comparation of MRM-MS method and LiBA method by measuring AFP and AFP-L3[59]
2.2 平行反应监测
近年来,高分辨质谱发展迅速,其具有扫描速率快、动态范围宽、定量精确等优点[61]。平行反应监测(PRM)是一种衍生于MRM的靶向蛋白质定量技术,具有Orbitrap质量分析器高分辨、高精度及四极杆高选择性的特点[62],已成为MRM的强大替代技术。与MRM相同,PRM首先以四极杆(Q1)作为过滤器来筛选目标肽段的前体离子,前体离子在Q2中碎裂之后,所有产物离子被收集在轨道阱(Orbitrap)中进行高分辨和高精度扫描,以获得MS2谱图[63]。由于PRM可以同时监控所有碎片离子,使其不再需要识别和验证特定离子,从而获得了更快、更简单的工作流程。PRM技术不仅具有SRM/MRM的定量分析能力,还具有定性分析能力,因此更适用于对蛋白质或多肽进行定性、定量分析[64-65]。Kim等[66]认为,PRM可以准确定量肺癌患者和健康对照者血浆中血清淀粉样蛋白A的特定亚型,在区分健康和疾病方面具有高精确度。Sathe等[67]通过使用高分辨质谱和串联质量标签(TMT)技术识别阿尔茨海默氏症的潜在脑脊液生物标志物,随后使用PRM对已知的潜在生物标志物进行验证,共识别出2 327个蛋白质,其中139个在阿尔兹海默症患者的脑脊液中变化明显。这些蛋白质有望用于监测疾病进展和药物治疗,一旦在大型研究中得到验证,还可用于阿尔兹海默症的早期检测。Hao等[68]为了验证用以区分髓母细胞瘤患者与对照患者的潜在尿蛋白生物标志物,采用基于TMT技术的定量蛋白质组学方法分别鉴定手术前后髓母细胞瘤患者和健康人群尿液蛋白质组中的差异蛋白,使用PRM方法在112个样本中验证了17种潜在生物标志物,其中CADH1、FGFR4和FIBB的组合可用于区分髓母细胞瘤患者和健康人群,这些发现将有助于尿液蛋白质组学在筛查和监测髓母细胞瘤中的应用,示于图7。

图7 通过PRM验证髓母细胞瘤尿蛋白生物标志物[68]Fig.7 Validation of medulloblastoma urine protein biomarkers by PRM[68]
2.3 数据非依赖采集
近年来,数据非依赖采集(DIA)技术备受关注,它是基于静电场轨道阱Orbitrap的一种全息式质谱数据采集模式[69],可以识别更多的肽段,且串联质谱后有更好的重复性、准确性及精密度[70-71]。在一级质谱检测后,DIA会对特定质荷比范围内的所有母离子进行碎裂,采集所有母离子的碎片离子,并快速依次扫描相邻母离子范围内的所有碎片离子,从而对蛋白进行定性和定量分析。DIA无需选择离子即可对所有离子进行检测,使其在较宽的质荷比范围内碎裂,提供更宽的检测动态范围,提高鉴定的重现性、灵敏度和准确性,有效地增强了蛋白质组的覆盖率,极大缩短了每个样品的检测时间,适合大样本的蛋白质组学研究;但DIA模式产生的碎片离子谱图过于复杂,可能丢失部分肽段及碎片离子的对应关系,对其结果的解析产生一定困难[72]。Rauniyar等[73]利用DIA及PRM技术证明载脂蛋白A-IV(ApoA-IV)可作为卵巢癌的肿瘤标志物,对卵巢癌的早检及靶向治疗具有积极意义。靶向卵巢癌蛋白质组分析表明,ApoA-IV作为卵巢癌肿瘤标志物的检测结果比免疫测定更可靠,示于图8。Song等[74]在LC-MS平台上构建了一个DIA工作流程,以揭示透明细胞癌(ccRCC)和相邻正常组织之间失调的蛋白质,实验发现436个蛋白质在ccRCC组织中失调,同时表明这些失调的蛋白质可能是ccRCC诊断的潜在生物标志物。

图8 在Thermo Fisher Scientific Orbitrap Fusion Tribrid质谱仪上采用DIA/PRM技术对实验组和对照组进行分析[73]Fig.8 Analysis of the experimental group and the control group by DIA/PRM technologyon the Thermo Fisher Scientific Orbitrap Fusion Tribrid mass spectrometer[73]
2.4 SWATH技术
SWATH是一种靶向DIA技术,其原理是将整个质谱扫描范围分割成多个区段,通过超高速扫描获得扫描范围内全部离子的所有碎片信息。本质上,MRM和PRM以前体离子为目标,仅收集目标离子碎片产物的信号,而SWATH-MS可以收集所有前体离子的信号,并通过提取目标肽段产生的数据实现靶向分析[75],其优势是降低了获得的片段信号的复杂性、具有优秀的重现性和准确性,在识别低丰度蛋白质时具有更高的灵敏度[76]。因此,SWATH技术在评估整个蛋白质组变化、分析特定的翻译后修饰(如磷酸化、糖基化等)以及建立蛋白-蛋白相互作用(PPI)动态图谱中有很好的表现[77]。但在使用SWATH-MS进行定量分析时,来自对照组的每个样本在质谱仪中单独运行期间的差异可能会影响生物标志物的相对定量和鉴定;较大的扫描宽度导致数据和噪声的复杂性增加、选择性显著降低,使得从复杂图谱中提取靶向肽段仍具有挑战性[78]。Chen等[79]运用SWATH-MS成功量化前列腺癌组织样本中的N-连接糖肽,以识别可能指示侵袭性前列腺癌的差异表达糖蛋白。Gao等[80]采用SWATH-MS方法对4 216个蛋白进行定量,示于图9,结果表明,HCC组织中有191个蛋白质上调,147个蛋白质下调,揭示了HCC中复杂的代谢过程,包括戊糖磷酸途径的改变、丝氨酸、甘氨酸和肌氨酸的生物合成/代谢、糖酵解、糖质新生、脂肪酸生物合成和脂肪酸β-氧化,为发现HCC新药物靶点或诊断性生物标志物提供了数据参考。

图9 肝癌组织与非肝癌组织的定量蛋白质组学分析[80]Fig.9 Quantitative proteomic analysis of HCC and non-HCC tissues[80]
3 结束语
对蛋白质类肿瘤标志物的准确定量有助于人们认识恶性肿瘤的发生发展机制,对恶性肿瘤的早期检测、良恶鉴别等具有十分重要的意义。近年来,生物质谱的快速发展极大推动了蛋白质类肿瘤标志物的研究进程。质谱技术在分辨率、质量精度和测序速度方面的发展为快速准确分析癌症蛋白质组提供了可能。但生物样品的高度复杂性使蛋白质类肿瘤标志物的准确定量存在很多困难,在临床中大范围地应用LC-MS技术定量分析蛋白质类肿瘤标志物还有待深入推进。开发高效快速的前处理方法及准确可靠的质谱扫描策略,从而发现更高灵敏度和特异性的肿瘤标志物,是推进恶性肿瘤早期检测以及制定个性化治疗方案的基础。随着仪器自动化技术的不断成熟,以症状为导向的诊疗方法将逐步向早期筛查和精准医疗转变,为恶性肿瘤患者提供新的生机。
