靶向小鼠ATP7B基因的CRISPR/Cas9系统构建与评价*
2022-06-15李欣怡孙雪梅岳鹏鹏于鸿浩
李欣怡,孙雪梅,付 灿,岳鹏鹏,于鸿浩,李 勇
(桂林医学院生物技术学院,广西 桂林 541199)
CRISPR系统是从细菌中发现的天然免疫系统,当病毒入侵时,该系统可改变外来基因组,以保护细菌细胞免受外来基因侵害。CRISPR/Cas9系统是由Ⅱ型CRISPR系统变化来的,使用CRISPR/Cas9技术可进行基因定向敲除、插入操作。与传统技术比较,其优势在于设计构建较为简单、操作简易、成本低、编辑效率高、可同时打靶多个基因等。基于CRISPR/Cas9的转基因技术已广泛应用于诸多领域,对人类产生了深远影响,有利于推动医药、育种等领域的快速发展[1-2]。
威尔逊病(WD)的全球患病率约为1/30 000,致病基因携带率约为1/90,在中国比较常见,是由铜转运P型三磷酸腺苷(ATP)酶基因突变引起[3]。WD的临床特点是代谢损伤会导致铜在各种组织(主要是肝脏和大脑)中的有毒积累,从而导致复杂的临床表现,如神经系统症状、肝硬化恶化、肾脏损伤、角膜色素环和其他症状[4-5],严重危害人类健康。本研究通过NCBI ClinVar等数据库比对分析,发现c.2333G>T(p.Arg778Leu)突变是中国人群突变的热点[6-7],故使用CRISPR/Cas9系统稳定编辑小鼠ATP7B基因c.2333G>T位点(p.Arg778Leu),建立高效、可模拟人ATP7B致病突变的小鼠ATP7B CRISPR/Cas9基因编辑系统,为进一步构建小鼠ATP7B基因突变动物模型奠定基础,也对精准模拟WD、深入理解WD的致病机制及探索可行的治疗策略具有重要意义。
1 材料与方法
1.1材料
1.1.1细胞株、菌株和质粒 大肠杆菌感受态菌株DH5α购于北京全式金生物技术(TransGen Biotech)有限公司;小鼠脑神经瘤N2a细胞由广州大学周建奎博士惠赠;pGL3-U6-sgRNA-PGK-puromycin (addgene#51133)和Cas9表达质粒pST1374-NLS-flag-linker-Cas9(addgene#44758)均由上海科技大学黄行许教授惠赠。T载体(pMDTM19-T Vector Cloning Kit)购于宝日医生物技术(北京)有限公司。
1.1.2试剂及仪器 TransDirect®Animal Tissue PCR Kit(AD201-01)购于北京全式金生物技术(TransGen Biotech)有限公司;去内毒素质粒中提试剂盒(CW2105)购于北京康为世纪生物科技有限公司;DNA纯化回收试剂盒购于天根生化科技(北京)有限公司;TaKaRa TaqTM(R001A)、Premix Taq均购于宝日医生物技术(北京)有限公司;Bsa Ⅰ购于NEB公司;Lipo fectamineTM3000 Transfection Reagent(InvitrogenTM)试剂盒、DMEM培养基、胎牛血清、杀稻瘟霉素和嘌呤霉素均购于赛默飞世尔科技公司;引物合成及DNA测序由北京擎科生物科技有限公司完成。实验所需的主要仪器包括超净工作台、台式离心机、CO2培养箱、聚合酶链式反应(PCR)仪、低温冷冻离心机、恒温摇床等。
1.2方法
1.2.1拟编辑位点的确定与sgRNA的设计 利用OMIM在线数据库筛查人ATP7B基因的功能失活突变,共筛查到25个突变位点(表1);为更广泛地筛查ATP7B致病突变,利用C1inVar数据库检索拼接位点突变的致病情况,发现变异位点多达300多个(表2)[8]。据研究可知,人ATP7B基因突变有明显的地域性,通过相关文献,找到在我国出现、明确、典型的致病突变,最终确定了人ATP7B.2333G>T(p.Arg778Leu)为拟突变位点。利用Ensembl数据库查询人ATP7B和小鼠ATP7B基因的结构,从Ensembl数据库中导出小鼠ATP7B基因序列,利用Vector NTI软件定位c.2333G> T位点序列,在其附近设计基因打靶位点,设计3个单向导RNA(sgRNA)序列。
1.2.2sgRNA表达载体的构建 设计好3个sgRNAs后,在各序列的5′端上添加ACCG合成正链,然后在其对应互补链的5′端上添加AAAC合成相应负链。将正负链退火得到黏性末端双链DNA片段。退火反应体系为:ddH2O 8 μL;正负链各5 μL(10 μmol/L),T7 Endonuclease I buffer 2 μL(10×),反应程序为:95 ℃ 5 min;95 ℃>85 ℃,共10个循环(每个循环降低1 ℃);85 ℃>25 ℃,共60个循环(每个循环降低1 ℃),-20 ℃保存。利用BsaⅠ酶对pGL3-U6-sgRNA载体进行酶切,得到产物pGL3-U6-sgRNA-BsaⅠ。连接酶切产物pGL3-U6-sgRNA-BsaⅠ和sgRNA的正负链退火后得到的具有黏性末端的双链DNA片段,将连接产物转化到大肠杆菌(DH5α)中,并涂布于LB固体培养基(氨苄抗性),在37 ℃环境下过夜培养18 h。用枪头在培养基上挑取单克隆,采用引物U6(通用)进行测序,鉴定阳性克隆。利用去内毒素中提试剂盒(CW2105 WBIO)提取出阳性克隆子的质粒,即pGL3-U6-ATP7B-sgRNA重组质粒。
1.2.3细胞转染及筛选 复苏小鼠N2a细胞,将之接种于DMEM完全培养基(含10% 胎牛血清),在5% CO2、37 ℃设定的培养箱中培养,每2~3天更换培养基,持续观察细胞状态。当其汇合度达到75%~85%时,用0.25%胰蛋白酶对细胞进行消化处理并传代至6孔板中,细胞悬液按比例分配,继续培养16~18 h,对比观察细胞形态、生长状况。当每孔内细胞汇合度达到70%~80%时进行细胞转染。利用LipofectamineTM3000 Transfection Reagen试剂盒将pST1374-NLS-flag-linker-Cas9质粒和相应的pGL3-U6-ATP7B-sgRNA质粒共转染小鼠N2a细胞,转染操作严格按照说明书进行。培养24 h后将转染液换成完全培养基,同时加入5 μg/mL嘌呤霉素和10 μg/mL杀稻瘟菌素进行药筛60 h,药筛后得到了阳性的sgRNA-Cas9共转染细胞。

表1 OMIM数据库中的ATP7B基因致病突变情况
1.2.4打靶位点处DNA的PCR扩增及测序、TA克隆 将药筛得到的阳性共转染细胞进行裂解,收集细胞裂解液,以细胞裂解液为模板对打靶位点处的目的DNA进行PCR扩增。PCR扩增反应体系为:细胞裂解液2 μL,上下游引物各1 μL;dNTP Mixture 2 μL,TaKaRa Ex Taq 1 μL、10×Ex Taq Buffer 2.5 μL;灭菌水加至25 μL。PCR扩增程序:95 ℃ 5 min;95 ℃ 20 s,57 ℃ 20 s,72 ℃ 25 s(共35个循环);72 ℃ 5 min,16 ℃无限时间。上述上游引物序列为5′-GGC AAG CGA CCC AGC AAG AAC C-3′,下游引物序列为 5′-TGC AGG CTG CCA GGA AAC TTC G-3′。扩增所得产物外送测序。将上步所得的PCR产物胶回收,得到的回收产物与pMDTM19-T载体连接,反应体系为:PMD19载体0.5 μL;Solution Ⅰ 2.5 μL;PCR纯化产物40 ng;加水补充至5 μL,16 ℃孵育1 h,得到连接产物。将所得产物快速转化到感受态大肠杆菌中,涂布于具有氨苄抗性的LB培养基(氨苄青霉素浓度为50 μg/mL)表面,37 ℃过夜培养20 h后,进行菌落PCR反应验证。根据电泳结果初步判断阳性克隆,再外送测序(Sanger)鉴定。
1.2.5打靶位点的基因型分析 针对打靶位点处的DNA序列与野生型DNA序列进行比对分析,在sgRNA3靶点位置发生了碱基的随机插入和缺失,记录每种改变情况并计算编辑效率。编辑效率=测序结果在打靶位点处发生碱基变化数/送测总数。

表2 ClinVar数据库中WD的ATP7B错义突变情况
2 结 果
2.1sgRNA表达载体构建成功 根据sgRNA符合5′-N(21)GG的原则,在ATP7B基因的打靶位点处设计3条sgRNAs,分别是sgRNA1(5′-TCA TGT GTT TTG AAT ATT GA-3′)、sgRNA2(5′-GCA TGC CGT CTA TTC TTA GT-3′)、sgRNA3(5′-GGA AAT ATA GGC CAA CTC CC-3′)。pGL3-U6-ATP7B-sgRNA 重组质粒的测序峰图(图1)中,选中部分为sgRNA的编码序列,与设计的3条sgRNAs一致,证明sgRNA表达载体构建成功。pGL3-U6-ATP7B-sgRNA重组质粒的凝胶电泳图,见图2。

A、B、C分别为pGL3-U6-ATP7B-sgRNA1、pGL3-U6-ATP7B-sgRNA2和pGL3-U6-ATP7B-sgRNA3质粒的测序峰图。
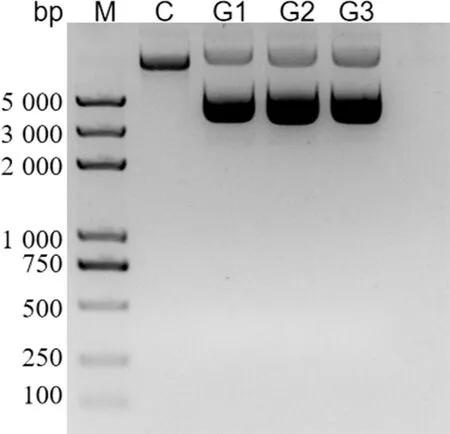
M、C、G1、G2、G3分别为Marker、Cas9、pGL3-U6-Galt-sgRNA1、pGL3-U6-Galt-sgRNA2和pGL3-U6-Galt-sgRNA3。
2.2获得sgRNA-Cas9阳性转染细胞 将pGL3-U6-ATP7B-sgRNA质粒和pST1374-NLS-flag-linker-Cas9质粒共转染小鼠N2a细胞,按照1.2.3方法成功筛选出sgRNA-Cas9阳性转染细胞,这些阳性转染细胞经过药筛作用后仍然正常生长,见图3。

A、B、C分别为药筛60 h后sgRNA1-Cas9、sgRNA2-Cas9和sgRNA3-Cas9阳性转染细胞图。
2.3sgRNA3导向序列打靶成功 将收集的阳性转染细胞裂解后作为模板,按照1.2.4方法进行打靶位点处DNA的PCR扩增,电泳结果(图4)显示,扩增条带清晰,片段大小也在预期的550 bp左右,可用于后续测序分析。测序峰图显示,只有sgRNA3靶点序列出现明显套峰(图5),初步说明sgRNA3导向序列打靶成功,靶位点发生了基因编辑;sgRNA1和sgRNA2靶点序列未出现明显套峰,说明这2个导向序列打靶不成功、靶位点未发生基因编辑或编辑效率较低。因此,后续实验只针对sgRNA3导向序列分析打靶效率。

M代表相对分子质量;NC表示野生型,G1、G2和G3分别为sgRNA1-Cas9、sgRNA2-Cas9和sgRNA3-Cas9转染型。

图5 sgRNA3打靶位点DNA的PCR扩增产物测序峰图
2.4sgRNA3打靶效率评价 按照1.2.5方法,利用TA克隆将sgRNA3靶位点DNA的PCR扩增产物连接到T载体上,通过菌落PCR初筛阳性克隆(图6),将阳性克隆子测序分析得到sgRNA3打靶位点的基因型(表3)。基因型分析结果显示,送测的克隆子部分发生了基因编辑,既有插入突变也有缺失突变,sgRNA3导向序列可以介导Cas9蛋白高效、特异性地切割靶点DNA,实现对小鼠ATP7B基因的编辑,编辑效率为53.85%[(4+1+2)/13]。

表3 sgRNA3靶点基因型分析

M代表Marker;1~15代表TA克隆的菌落序号。
3 讨 论
WD患者如不能及时进行积极治疗,至晚期则因严重肝硬化、肝功能衰竭或并发感染而死亡[9]。在1948年以前,WD因无有效疗法,病程大多为3~4年。WD的发病率为1/100 000~1/30 000,在中国较多见,且多见于青少年[10]。目前,已有学者建立人ATP7B转基因大鼠模型,并对转基因大鼠体内的表达分析进行研究[11]。当前的医疗手段并不能完全治愈WD,只能通过长期控制饮食、服用药物来维持体内酮含量水平正常[12-15]。目前,大部分WD相关研究仍然集中在蛋白分析或临床研究上,少见有关基因突变细胞模型、动物模型建立的相关研究,这在很大程度上制约了国内WD研究的进一步深入[16]。
本研究主要通过使用CRISPR/Cas9系统在N2a中稳定编辑ATP7B基因,建立WD致病突变的细胞模型,为其分子机制的更深入研究与基因治疗打下前期基础。本研究基因型分析结果显示,部分克隆子发生了基因编辑,sgRNA3导向序列能够介导Cas9蛋白对目标DNA的高效特异性切割,编辑效率为53.85%,但sgRNA1和sgRNA2没能有效结合Cas9蛋白完成基因编辑,作者推测可能是这2种预设的sgRNA不能有效介导Cas9蛋白切割基因组,以后可能通过优化sgRNA设计解决[17],亦可尝试使用保真度更高的核酸酶替代[18]。
本研究采用TA克隆方法分析基因型和敲除效率,此方法检测快速,对样品数量要求低,但使用的CRISPR/Cas9系统也面临许多挑战,例如非目标效应或DNA的大量破坏和重排,如果修复不当,CRISPR/Cas9介导的突变会诱导异常发育等[19-20]。仍然需要进一步开展对CRISPR/Cas9靶向ATP7B非目标效应相关研究。
综上所述,本研究成功采用CRISPR/Cas9系统进行小鼠ATP7B基因的细胞水平编辑,编辑效率为53.85%,这为更深入开展WD的分子机制研究和疾病干预奠定了科学基础。
