分子识别特征预测算法特性分析*
2022-06-13李子夏
李 子 夏
(天津大学应用数学中心,天津 300072)
0 引 言
固有无序蛋白区域(intrinsically disordered proteins regions,IDRs)在生物体中占有重要地位.具有IDRs的蛋白质能影响分子组装识别、信号传导、重排、转录和翻译等细胞功能[1],并参与小分子的结合、转运和催化[2].Habchi等[3]证实约30%~50%的真核蛋白具有一个或多个长的IDRs.固有无序蛋白结合域是IDRs的功能区域,在细胞的信号传递和调节过程中起着重要作用,是无序蛋白质研究的热点.固有无序蛋白结合域按照区域的长短分为短线性基序(short linear motifs,SLiMs)和分子识别特征(molecular recognitionfeatures,MoRFs),其中SLiMs和MoRFs的残基数分别是≤5和5~25个.Yan等[4]分析了868个完整蛋白质组,结果显示真核生物有21%的IDRs具有MoRFs,细菌和古细菌有29%的IDRs具有MoRFs.
由于SLiMs和MoRFs长度上的差异,所以预测这2类功能域的方法不同.目前SLiMs的预测是基于在一组不同序列中寻找正则表达式的原理来开发算法.MoRFs相比于其他无序区域和结构化区域有其独特的序列特征,因此,MoRFs的预测可以基于序列进行精确的计算预测.另外,MoRFs的长度越长,序列特征越明显,如:与其他IDRs相比,MoRFs区域富含大的疏水侧链的氨基酸,特别是芳香族氨基酸含量较高;与SLiMs的预测算法相比,MoRFs的预测算法更多,准确率也更高.这些预测算法的出现,推动了MoRFs计算识别算法的发展[4],推定MoRFs不仅有助于阐明蛋白质功能,还可用于多种病毒蛋白质组、细胞死亡途径、通道蛋白的相互作用组、激酶、核小体和核糖体的分析研究[5-10].
近年来,研究人员相继提出一批基于不同原理及方法的MoRFs预测算法[11-12].Dosztanyi等[11]基于多肽链中残基的3种性质,结合残基必须处于一个长的无序区域、残基不能与其领域折叠以及残基能够与球状结合域相互作用,开发了ANCHOR;Malhis等[12]利用贝叶斯规则结合了MoRFs的保守性、MoRFs与其侧翼IDRs的理化性质差异以及其本身的无序特征,开发了MoRFCHiBi_web.然而,经过生物学家实验确认的MoRFs数量很少,只有53条蛋白质序列中包含MoRFs[12],因此MoRFs预测算法之间有很大差别.近年来,随机序列已被广泛应用于生物信息学多个领域的算法对比分析研究中[13-14],因此本文构造随机蛋白序列作为测试集,将其创造性地应用于MoRFs的预测算法比较分析中,选取2种经典的MoRFs预测算法比较其结果差异与特性,以期将来对MoRFs更深入的研究建立理论基础.
1 材料与方法
1.1 构建数据集
通过产生随机序列作为独立数据集,将20种氨基酸随机排列,得到随机序列,规定每种氨基酸的使用频率为5%.通过等比例随机取样的策略,从固定的20种氨基酸残基的组合中,即丙氨酸(A)、精氨酸(R)、天冬酰胺(N)、天冬氨酸(D)、半胱氨酸(C)、谷氨酰胺(Q)、谷氨酸(E)、甘氨酸(G)、组氨酸(H)、异亮氨酸(I)、亮氨酸(L)、赖氨酸(K)、蛋氨酸(M)、苯丙氨酸(F)、脯氨酸(P)、丝氨酸(S)、苏氨酸(T)、色氨酸(W)、酪氨酸(Y)和缬氨酸(V)),随机生成10 000条长度均为60个残基的随机蛋白序列,接下来使用CD-HIT工具(相似度阈值参数设定为30%)[15],对这些蛋白质序列去冗余.以此数据集进行实验验证,将其记为Rseq.
1.2 MoRFs预测算法
选取ANCHOR和MoRFCHiBi_web算法进行对比分析.ANCHOR被嵌入到MobiDB3.0数据库中用于预测MoRFs,是一个非常经典的算法;MoRFCHiBi_web是2016年被开发出来的算法,比之前开发的其他MoRFs预测算法准确率高.其中,MoRDCHiBi_web算法比ANCHOR的计算时间长,因为MoRDCHiBi_web算法为了计算保守性特征需使用 PSI-BLAST工具[16].
1.3 评估指标
1.3.1 氨基酸类型偏好
统计数据库中每种氨基酸在MoRFs区域或非MoRFs区域上出现的频率与其在整个数据集中的出现频率之差,来表示MoRFs区域中各种氨基酸的使用偏好,公式如下:
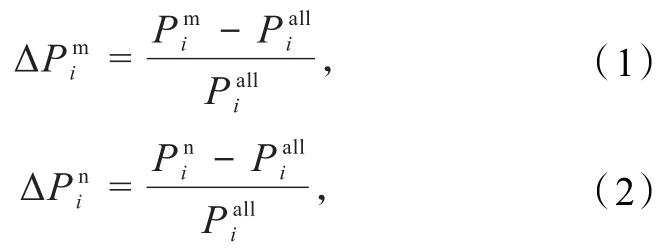
除此之外,将每一条序列分为MoRFs区域、Flanks区域(MoRFs两侧各含8个残基)和Others区域(除MoRFs和Flanks区域外)共3个区域,统计这3个区域的氨基酸类型偏好.
1.3.2 相关性分析
使用Pearson相关系数(r)衡量2种预测方法预测结果的相关程度,将数据集Rseq中的随机序列随机等分为10组,每组的全部残基在2种方法的预测概率分数分别构成一个向量,对其进行相关性分析.相反系数计算公式为
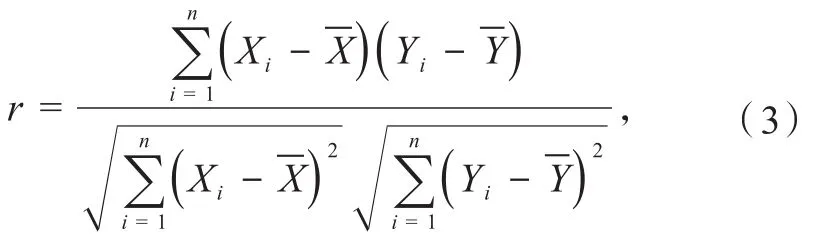
式中:r∈[-1,1],r>0为正相关,r<0为负相关,|r|越大则相关性越高.
1.3.3 平均得分
统计分析2种算法预测每条序列时,每个残基的概率值与其在序列中的位置关系.计算Rseq上从1~60的每个位置上残基得分的算术平均数(pavg),计算公式为

式中:pi表示第i条序列上某个位置上的预测得分值,n为序列的总个数.当算法在所有位置上的平均得分都相近时,表明该算法对每个残基预测得分与残基所在位置无关,否则表明该算法对残基预测得分与残基所在位置有关.
2 结果与分析
2.1 预测结果比较
ANCHOR算法预测时,10 000条序列均返回预测结果;MoRFCHiBi_web算法预测时,有9 271条序列返回预测结果.MoRFCHiBi_web算法使用了PSI-BLAST用以计算序列保守性特征,当序列在此过程中找到同源序列才得以计算后续特征,否则不能得到预测结果.ANCHOR和MoRFCHiBi_web算法对Rseq的随机蛋白序列预测的总无序残基和与其对应的蛋白质序列数目的关系如图1所示.2种算法都预测出了MoRFs残基,并且都有大量的序列没有被预测到正样本.ANCHOR和MoRFCHi-Bi_web分别在5 628和4 595条序列上均预测为非MoRFs残基.ANCHOR能够预测到的一条序列上的MoRFs残基数大多集中在1~8个,MoRFCHi-Bi_web预测到的一条序列上的MoRFs残基数大多集中在0~10个.与ANCHOR算法相比,MoRFCHi-Bi_web算法预测的MoRFs残基长度整体偏大.
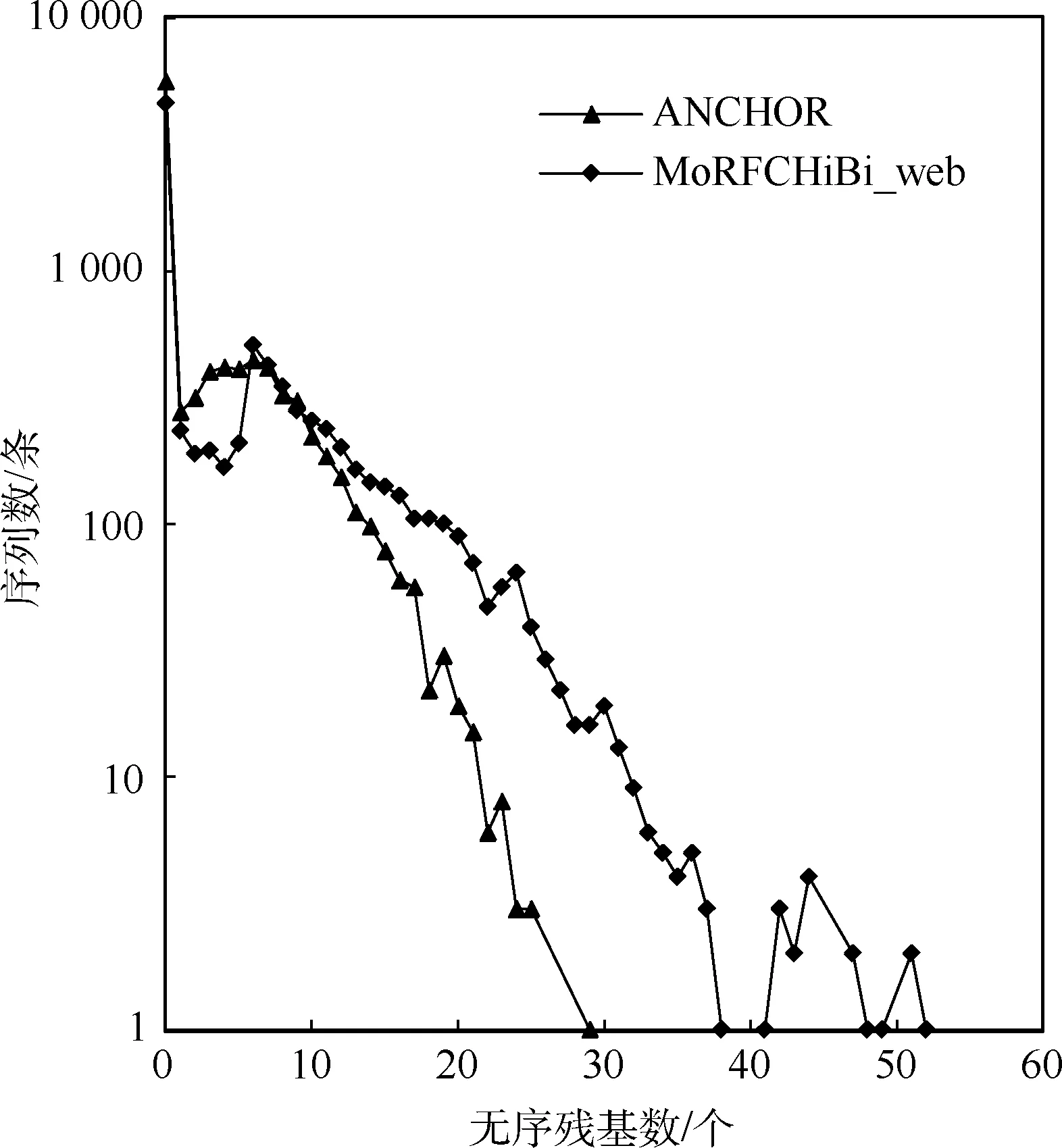
图1 2种算法对Rseq中不同总MoRFs残基长度上的序列分布
2.2 氨基酸类型偏好分析
ANCHOR和MoRFCHiBi_web算法预测的氨基酸类型偏好结果如图2.可知2种算法得到的结果整体一致,MoRFs区域上偏好的氨基酸类型有10种,分别是A、R、I、M、L、F、P、W、Y和V;非MoRFs区域上偏好的氨基酸类型也有10种,分别是N、D、C、Q、E、G、H、K、S和T.这与Yu等[13]的研究结果略有不同,其研究显示固有无序区域和有序区域上偏好的氨基酸类型分别有12和8种,无序区域偏好的氨基酸类型为A、R、N、D、Q、E、G、H、K、P、S和T,有序区域偏好的氨基酸类型为C、I、L、M、F、W、Y和V.MoRFs区域偏好的氨基酸类型与无序区域上偏好的氨基酸类型有很多的区别,这是由于MoRFs区域相对于其他无序区域有结构化的趋势,氨基酸类型使用偏好上会有结构蛋白的特征.
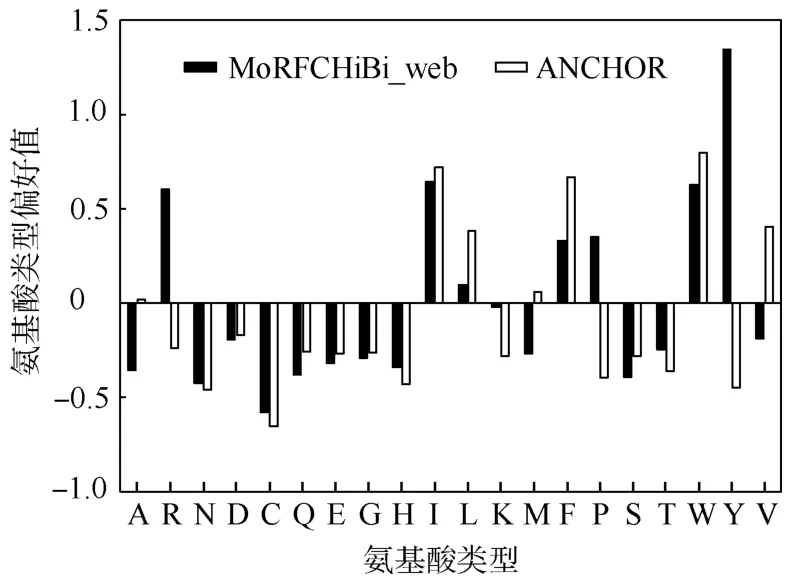
图2 在MoRFs区域上2种算法预测的氨基酸类型偏好
2种算法预测的3个区域的氨基酸类型偏好结果如图3所示.ANCHOR算法预测的MoRFs区域偏好的氨基酸类型有6种,分别为A、I、L、M、F、W和V;Flanks区域偏好的氨基酸类型有5种,分别为A、Q、E、I和V;其在Others区域偏好的氨基酸偏好类型有2种,分别为C和Y.MoRFCHiBi_web算法预测的MoRFs区域偏好的氨基酸类型有6种,分别为R、I、F、P、W 和 Y;Flanks区域偏好的氨基酸类型有8种,为A、N、D、Q、E、G、K和S;其在Others区域偏好的氨基酸类型有3种,分别为C、H和V.比较可知,MoRFs和Flanks区域上的氨基酸类型偏好值均较大,Others区域上的20种氨基酸类型偏好值均较小.
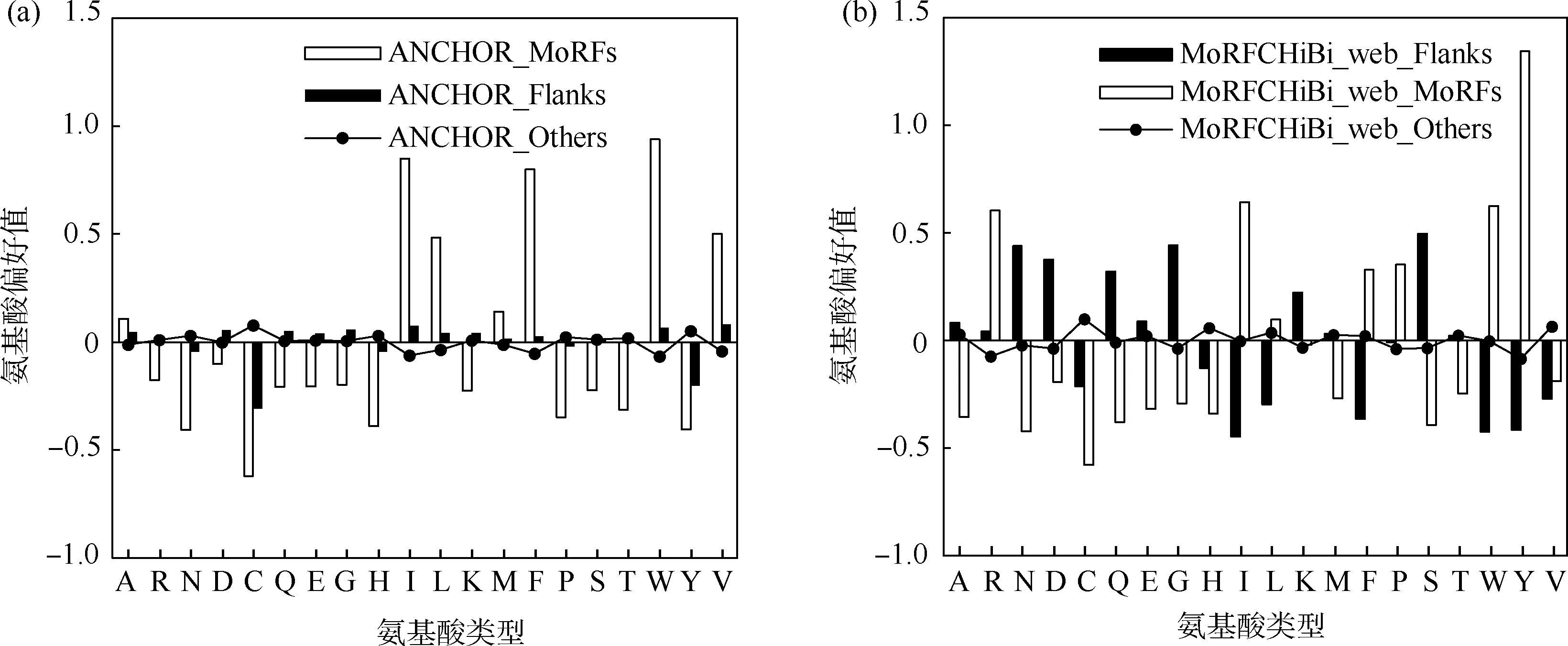
图3 不同算法在3个区域上预测的氨基酸类型偏好
2.3 预测算法相关性分析
10组向量的r分布在0.19~0.25,r的平均值为0.21,说明2种算法的预测存在正相关,但相关性较低.本文结果与Yu等[13]的分析结果不同,说明相对于蛋白质无序的预测,MoRFs的预测难度要更大,MoRFs预测算法的一致性更低.
2.4 平均得分
Rseq上从1~60的每个位置上残基的平均得分与位置分布如图4所示.ANCHOR和MoRFCHi-Bi_web算法预测残基的60个位置的平均得分分别为 0.162~0.176和 0.576~0.613.可知,ANCHOR算法对残基预测的平均得分与其在序列中的位置几乎没有关系,MoRFCHiBi_web算法预测残基的平均得分与残基所在的位置有较明显的关系,即序列两端位置的残基平均得分更高,序列中间位置残基平均得分更低,表明序列两端位置的残基更容易被预测为MoRFs.
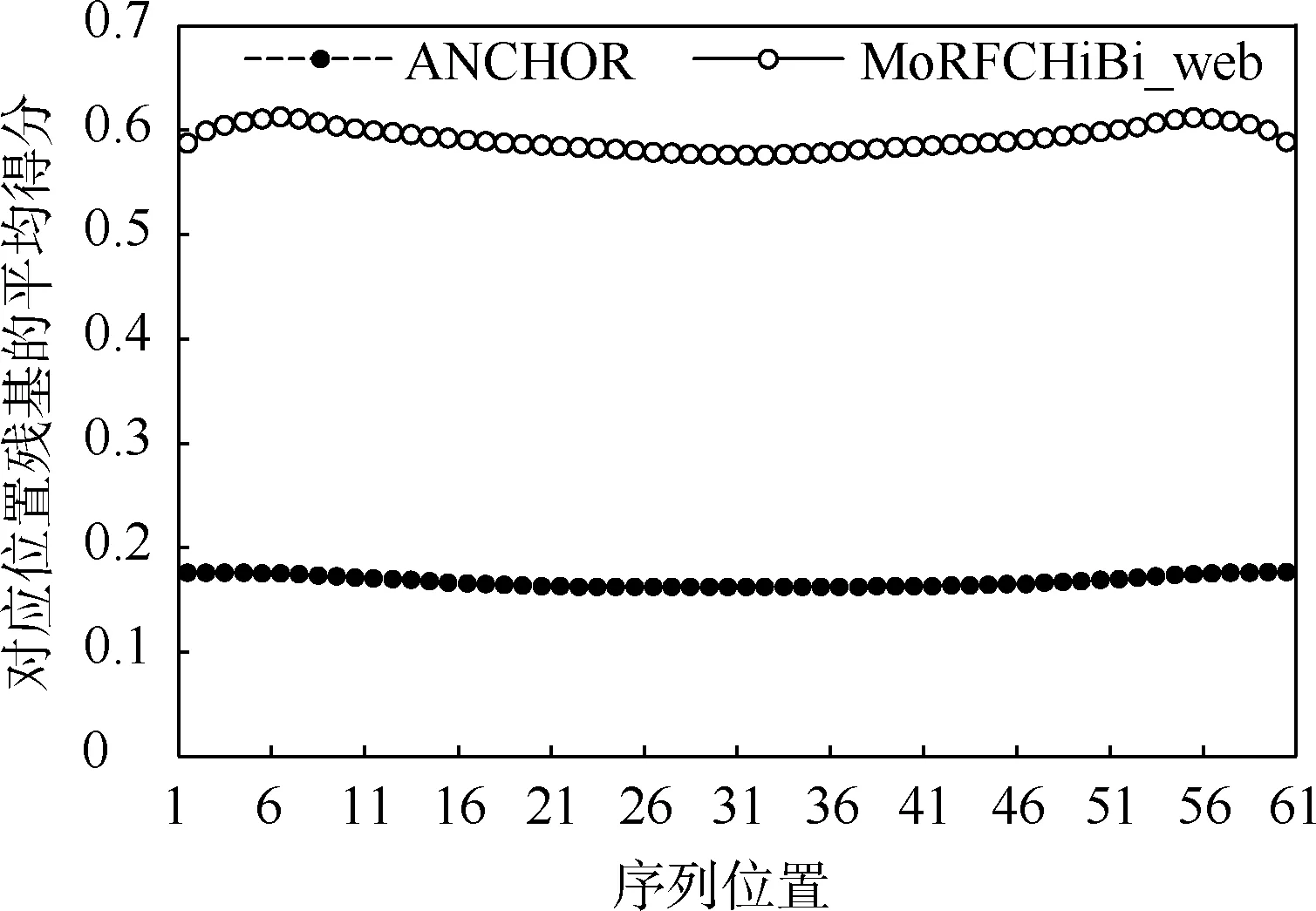
图4 残基所处位置与平均概率的关系
3 结束语
本文基于随机蛋白序列,系统地对比分析了2种MoRFs预测算法在数据集Rseq上的预测结果.MoRFs残基分布的分析表明,与ANCHOR算法相比,MoRFCHiBi_web算法预测的MoRFs残基长度整体偏大;氨基酸类型偏好分析表明,MoRFs虽然是无序区域的一部分,但MoRFs上的氨基酸类型偏好与无序区域的氨基酸类型偏好有很大的差别,MoRFs与Flanks区域的氨基酸偏好较为明显,这为MoRFs的研究提供了新的思路;就预测的位置而言,MoRFCHiBi_web算法在序列两端的残基更容易被预测为MoRFs,但ANCHOR算法预测的结果与残基所处位置基本没有关系.综上,2种方法的预测结果存在差异.因此,为了提升实际工作效率,科研人员应根据实际需要选择不同的MoRFs预测算法.
