有机相生物传感器电催化反应机理及其在植物油检测中的应用
2022-06-11于丽敏
陶 莎,于丽敏
(1.中国农业大学 信息与电气工程学院,北京 100083;2.山东农业工程学院信息科学与工程学院,山东 济南 250100)
1 引言
过氧化物在化学、医药、生物、食品、环境等行业广泛应用与存在,过氧化氢以及其它有机过氧化物对人体具有严重的危害,主要表现为:进入人体内转化为过氧化自由基,可激活酪氨酸氧化酶或直接催化酪氨酸产生黑色素,加速机体的衰老[1];自由基可通过损伤DNA导致肿瘤的发生[2];可导致血脂增高,血管内壁增厚,最终导致动脉粥样硬化的发生[3],因此准确、快速测定食品、环境、医药中的过氧化物含量水平具有重要的现实意义。
采用过氧化物酶电极法分析过氧化物是近年来研究的热点,已广泛用于生化、环境、食品等领域,但研究大多集中于水相介质[4,5]。制约酶电极在有机相中应用,主要是由于酶电极在有机相溶剂中的电化学催化反应特性与水相中的酶催化反应是完全不同的,其催化反应机制要复杂得多,涉及到固定化酶存在形式、固定化载体的状态、酶与底物之间催化反应机理、电极表面与酶活性中心之间的电子传递速率、底物在有机相和酶修饰膜层的传质动力学、有机溶剂本身性质等诸多因素。其中酶的固定化方法是影响有机相酶电极性能的关键因素,一般酶的固定化方法包括共价键合法、共价交联法、吸附法、掺杂法、电化学聚合法等[6-9],前两种方法不适合有机相酶电极的酶固定化,因为有机溶剂会攻击共价键,使其变性或脱落,而吸附法简单可行,但该法酶的吸附固定量小,泄漏比较大,造成酶电极的响应信号小,稳定性不足。
溶胶-凝胶(Sol-Gel)材料的提出,促进了有机相生物传感器的发展,因为该材料具有其它高分子材料无法比拟的特性[10],如较好的物理刚性,化学惰性及透明性,良好的机械稳定性,可忽略的溶胀性,其可调的多孔网状结构能使酶在较低的温度下被包埋固定而且不易泄漏,较好的生物相容性有利于保持酶自身的结构、活性和功能,同时小分子底物可以通过其微型孔道与酶接触反应,可以为微型网格中的生物活性物质如酶、抗体、细胞提供良好的酶反应微环境,所以已成为非水酶学具有重大应用价值的领域之一。
采用溶胶-凝胶固定法制备的电极具有响应快,固定酶量大等特点。刘俊女[11]等采用溶胶-凝胶法包埋酪氨酸酶,制备酶修饰碳糊电极,并将其应用于酚类物质的检测中,结果对邻苯二酚、对邻甲酚、对苯二酚等酚类有良好响应。李彤[12]等基于普鲁士蓝修饰玻碳电极结合二氧化硅溶胶-凝胶固定化酶技术,构造具有“三明治”式结构的酶电极。结果表明,制备得到的酶电极对葡萄糖在0~5mmol/L范围内呈线性响应。目前此法制备得到的酶电极在水溶液体系中能够很好的检测,稳定性及灵敏度均优于其他固定法,但由于有机相中物质的传递速率与水相中的不同,其能否用于有机相酶电极的构造仍有待研究。
本研究拟以溶胶-凝胶方法制备辣根过氧化物酶(HRP)电极,通过试验确定最佳的技术参数,以构建良好的酶促反应微环境和较高的电子传递速率,制备出稳定性好、响应灵敏的HRP酶电极;根据HRP酶的生物学特性和有机过氧化物的性质选择溶解性好、稳定性好、电化学性能适宜的有机溶剂或复合有机溶剂及有机电解质以构建HRP酶电极催化反应的有机介质体系;利用各种结构分析方法和手段考察HRP酶在有机相介质中其构象、活性变化规律,揭示HRP酶对有机过氧化物的催化反应过程,建立酶电极电催化反应的动力学模型;本研究通过酶学、生物化学、材料科学和电化学知识的综合应用有助于促进有机相酶电极对有机过氧化物电催化还原规律的认识,推动有机相酶电极在测定有机过氧化物及其它非水溶性微量有机物质中的应用,尤其是在食品安全和生物体系中过氧化脂质的测定方面,为进一步开发工业化应用的生物传感器及相关仪器提供基础。
2 材料与分析方法
2.1 试剂与材料
过氧化月桂酰(95%):美国Fluka公司;辣根过氧化物酶:北京拜尔迪生物技术有限公司;十六烷基三甲基溴化铵(CTAB):Amresco,分析纯;磷酸二氢钠、磷酸氢二钠、氯化锂、硫酸、乙腈、1,2-二氯乙烷、碘化钾、硫代硫酸钠、可溶性淀粉、水杨酸、冰乙酸、异辛烷均为分析纯,北京化工厂;植物油样品:海淀区产品质量监督检验所从北京市内市场、工厂、餐饮店等收集得到。
2.2 实验仪器
电化学工作站 (CHI650C)、玻碳电极(CHI114)、 铂丝电极 (CHI 104)、Ag/AgCl电极(CHI 111)、电极抛光材料(CHI120):均为上海辰华仪器公司;超声波清洗器(KQ100E):昆山市超声仪器有限公司;电子分析天平(AR2140):奥豪斯仪器(上海)有限公司;微量移液器:Finnpipette雷勃移液器;磁力搅拌器(GL3250A):海门市其林贝尔仪器制造有限公司。
2.3 实验方法
2.3.1 辣根过氧化物(HRP)酶电极的制备
玻碳电极依次用 1.0、0.3、0.05 μm 的 Al2O3粉研磨抛光,用超纯水润湿,接着进行超声清洗处理,再用超纯水和丙酮冲洗除去其表面吸附物质,使成镜面,然后在0.2 mol/L的硫酸溶液中用循环伏安法扫描处理,扫描速度为50 mV/s,电压范围为-0.5~+1.5 V,持续扫描10 min,得到稳定的循环伏安图后,取出用二次蒸馏水洗净,室温干燥后备用。
取150 μL正硅酸乙酯与100 μL水依次加入到400 μL甲醇溶液中,混匀后加入50 μL亚铁氰化钾溶液、5 mmol/L NaOH溶液 25 μL和5%CTAB 甲醇溶液 25 μL,超声震荡 1 min,然后吸取15 μL滴加到清洗干燥后的玻碳电极上,室温下干燥使溶剂蒸发,在电极表面形成一层掺杂了亚铁氰化钾的凝胶膜。
接着取 150 μL 正硅酸乙酯、100 μL 水、5 mmol/L NaOH溶液25μL及5%CTAB甲醇溶液25 μL依次加入到400 μL的甲醇溶液中,混匀后加入50 μL HRP酶溶液,超声震荡30 s,然后吸取15 μL滴加在干燥后的亚铁氰化钾修饰电极上,室温下放置,待溶剂蒸发后,即形成HRP酶电极,制备好的酶电极在4℃下保存备用。
2.3.2 电化学分析技术
本研究采用循环伏安法和计时电流法研究HRP酶修饰电极在有机相中的电化学行为,并对油脂中过氧化物的催化反应及电极过程进行分析;通过循环伏安法测量修饰电极对油脂中过氧化物的电流响应,得到电流-电位曲线。
2.3.3 标准溶液的配制及检测
准确称取0.4 g过氧化月桂酰标准品,加入到100 mL容量瓶中,用1,2-二氯乙烷定容,之后再用1,2-二氯乙烷准确稀释500倍,作为标准贮备液避光贮存备用,此时过氧化月桂酰浓度为8μg/mL。取5支25 mL容量瓶,按照表1中所述的量分别加入过氧化月桂酰标准贮备液、1,2-二氯乙烷、0.1 mol/L硫酸水溶液及1.0 mol/L氯化锂乙腈溶液,最后用乙腈定容至25 mL,作为过氧化月桂酰标准工作液待测,此时过氧化月桂酰最终浓度分别为 0.32、0.64、0.96、1.28、1.6μg/mL。
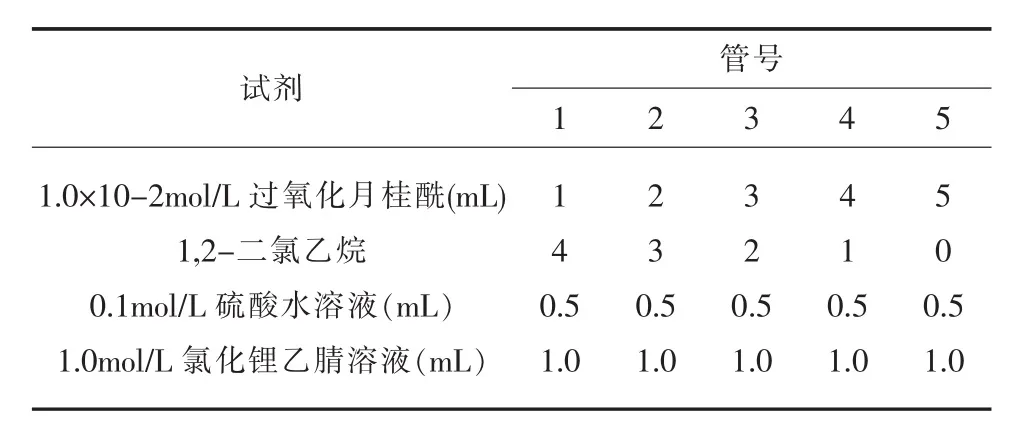
表1 不同过氧化月桂酰浓度配制方法Table1 The method of preparation for different lauroyl peroxide level
之后采用线性伏安法,使用HRP酶修饰电极在0~0.7V电位下,以70mV/s的速度记录电流-电位曲线,同时用该电极做空白溶液的线性伏安图,每个样品平行测量三次,取其峰电流值的平均值,并在此基础上减去电极在空白溶液中产生的峰电流值,以此差值作为过氧化月桂酰的电流响应。再以过氧化物浓度为横坐标,该电流响应为纵坐标绘制标准曲线,建立回归方程。
2.3.4 植物油样品的检测
准确称取植物油样品1.00 g于25 mL容量瓶中,加入5 mL 1,2-二氯乙烷使之充分溶解,再加入0.1 mol/L硫酸水溶液0.5 mL及1.0 mol/L氯化锂乙腈溶液 1.0mL,最后用乙腈定容,即为植物油样品待测液。接着按照2.3.3中所述方法采用HRP酶修饰电极进行线性伏安分析,记录电流-电位曲线,将得到的峰电流值扣除空白溶液的峰电流值后,根据2.3.3中得到的回归方程,推算出该植物油样品的过氧化物含量,再换算成相应的过氧化值。
最后将采用HRP酶修饰电极法得到的结果与采用国标碘量法测定得到的结果进行比对,判断两种方法所得结果是否一致。
3 结果与分析
3.1 亚铁氰化钾的添加量对修饰电极峰电流的影响
为了提高酶电极的催化反应速率及响应电流,本项目选用含有电子介体的酶电极,即在第一层凝胶膜中掺杂亚铁氰化钾,让其在HRP酶与电极之间起到传递电子的作用。实验分别配制了不同浓度的亚铁氰化钾溶液(5.5、6、6.5、7、7.5、8、8.5、9、9.5、10mmol/L),以亚铁氰化钾浓度为横坐标,还原峰电流响应为纵坐标作图,结果如图1所示。
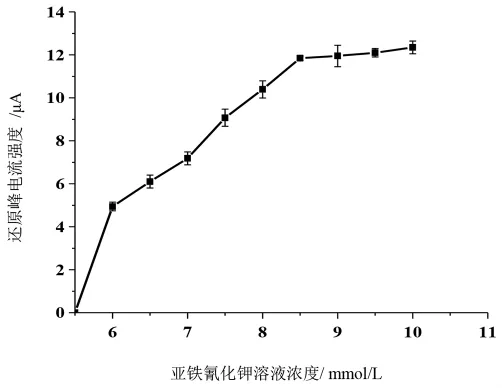
图1 亚铁氰化钾添加量对还原峰电流响应的影响Fig.1 The influence of adding amount of potassium ferrocyanide on the reduction of peak current
由图可知,随着亚铁氰化钾浓度的上升,酶电极还原峰电流响应值逐渐升高。而亚铁氰化钾同时还会对凝胶膜特性产生一定影响。由于电解质的加入会引起溶胶黏度的增加并促使溶胶快速转变为凝胶,当亚铁氰化钾的浓度较低时(10mmol/L以下),溶胶母液表现出“触变性”,即形成的凝胶经振动,便等温可逆的转变为溶胶;静置后又成为凝胶,这是由于在外力作用下体系的黏度减小,流动兴变大造成的。此时的溶胶是可用于制备酶电极的,但随着亚铁氰化钾添加量的不断增大(10mmol/L以上),这种转变将不再可逆,而是表现出离浆特性,即形成的凝胶从溶剂中脱离出来,沉在试管底部,此时的母液将不能用于修饰膜的制备。因此,本研究选择亚铁氰化钾浓度为10mmol/L的溶液进行酶电极的制备。
3.2 HRP酶的添加量对峰电流的影响
为了研究HRP酶添加量对酶电极催化过氧化月桂酰产生的还原电流响应的影响,本试验分别配制了不同浓度的HRP缓冲液,依次为6、7、8、9、10、11、12 mg/mL 按 2.3.1 中所述的程序制备7支酶电极,分别记录其在0.8μg/mL过氧化月桂酰溶液中的还原电流响应,并绘出还原峰电流响应强度与HRP酶浓度间的关系曲线,结果如图2所示。

图2 HRP酶添加量对还原峰电流响应的影响Fig.2 The influence of adding amount of HRP enzyme on the reduction of peak current
在一定范围内,HRP酶电极对过氧化月桂酰的电流响应随着HRP酶浓度的增加而逐渐增大,当酶浓度超过10 mg/mL以上时,其电流响应的增加趋势逐渐趋于平缓。究其原因当酶浓度较低时,固定在电极上的HRP酶量随着缓冲液中HRP酶浓度的增加而增加,产生的催化还原电流响应值也会随之增大;而当缓冲液中HRP浓度过高(高于10 mg/mL)时,修饰电极表面固定的HRP酶量与电极表面接触到的过氧化月桂酰的量相比,处于过剩状态,此时再提高酶的浓度,其电流响应的增加趋势会逐渐趋于平缓。因此,综合油脂过氧化物的测定范围考虑,本试验选择10mg/mL为最佳HRP浓度。
3.3 HRP酶修饰电极在有机相中的电催化反应机理
3.3.1 HRP酶修饰电极在有机相中的伏安行为
为了研究采用溶胶-凝胶法制备的HRP酶修饰电极在有机相中的电化学行为,分别记录了未经修饰的裸玻碳电极与经过修饰的HRP酶电极在不同溶液中的循环伏安曲线。图3(1)为裸玻碳电极在乙腈-1,2-二氯乙烷混合溶液 (体积比为4:1,并添加了0.1mol/L硫酸水溶液0.5mL及1.0mol/L氯化锂乙腈溶液1.0mL)中的循环伏安图,由图可以看出裸玻碳电极在扫描电位-0.7~0.7V范围内,没有出现任何峰电流,说明裸电极在有机相介质中既无氧化作用也无还原作用发生。
图3(2)为经HRP酶修饰的玻碳电极在相同溶液中的循环伏安图。图中显示,HRP酶修饰电极在0.598V及-0.294V处分别形成了一个氧化峰和一个还原峰,虽然峰形较好,但氧化峰与还原峰之间的电位差△Ep≈892mV。

图3 裸玻碳电极(1)与修饰电极(2)在乙腈/1,2-二氯乙烷混合溶液中的循环伏安图(电解液25mL,乙腈/1,2-二氯乙烷体积比4:1,并含有0.1mol/L硫酸水溶液0.5mL及1.0mol/L氯化锂乙腈溶液1.0mL,扫描速度30mV/s)Fig.3 Voltammograms of the bare GC electrode(1)and HRP enzyme modified electrode(2)in the mixed solution of acetonitrile-1,2-dichloroethane(the volume ratio was 4:1,and 0.1 mol/L aqueous sulfuric acid solution of 0.5 mL and 1.0 mol/L acetonitrile solution of lithium chloride of 1.0 mL were added),at the scan rate of 30mV/s.
A.J.Bard[13]认为,当循环伏安图中出现一对峰形良好的氧化还原峰时,如果其峰电位差△Ep在100mV以内,可认为反应体系是可逆的;如果其△Ep在200mV以内则说明体系准可逆;如果△Ep大于200mV或者只有一个峰出现时,则判定体系完全不可逆。
图3(2)中的氧化峰与还原峰之间的电位差明显大于200mV,说明HRP酶电极在该有机相体系中是完全不可逆的,即有机相体系中的亚铁氰化钾/铁氰化钾电子对在HRP酶与电极表面传递电子的速度缓慢,这一点也可通过其峰电位时常出现位移这一现象得到证明。
通常,在可逆体系、准可逆体系中,峰电流的波动较大,而峰电位一般不发生改变,而在不可逆体系中,峰电流在波动的同时,峰电位也会发生位移化,甚至波动的范围比峰电流还大。在本体系中,在用伏安法测定峰电流及峰电位时,峰电位时常会出现位移,变异系数在0.025~0.062(n=19)。这与判断该反应体系是完全不可逆的结论相吻合。
为了证明是有机相引起的电子间传递速率下降,采用HRP酶修饰电极在PBS缓冲溶液中进行循环伏安分析,结果如图8所示,在扫描电位0~0.4V范围内出现了一对峰形良好的氧化还原峰,且它们之间的峰电位差△Ep≈36mV,氧化峰电流与还原峰电流值之比i(pa)/i(pc)≈0.75,说明HRP酶电极在水相体系中是可逆的,即在水相体系下亚铁氰化钾/铁氰化钾电子对能有效地在电极与HRP酶表面传递电子。

图4 HRP酶修饰电极在0.2mol/LPBS缓冲溶液(pH6.8)中的循环伏安图(扫描速度30mV/s)Fig.4 Voltammograms of HRP enzyme modified electrode in the 0.2mol/L PBS buffer solution(pH6.8),at the scan rate of 30mV/s
为了提高有机相中亚铁氰化钾/铁氰化钾电子对在酶与电极间的电子传递速度,逐渐提高扫描速度,发现在一定范围内随着扫描速度的提高,氧化峰与还原峰之间的电位差在逐渐缩小,且当扫描速度在50~75mV/s时,峰电位差小于200mV,其中当扫描速度在 70mV/s(图 5)时,峰电位差为170mV,氧化峰电流与还原峰电流值之比i(pa)/i(pc)≈0.83,此时为准可逆体系,考虑到当扫描速率过快时,电极会快速极化,反应体系达到的可能是暂态状态而不是稳态,因此在本试验中,HRP酶修饰电极在有机相介质中的扫描速度采用70mV/s。

图5 HRP酶修饰电极在有机相中的循环伏安图(扫描速度70mV/s)Fig.5 Voltammograms of the HRP enzyme modified electrode in the mixed solution of acetonitrile-1,2-dichloroethane,at the scan rate of 70mV/s
3.3.2 HRP酶修饰电极对过氧化月桂酰的催化反应机理
为了研究HRP酶修饰电极对脂质过氧化物的催化作用,除记录裸玻碳电极及酶修饰电极在空白有机相介质中的循环伏安图外,增加记录了裸电极及酶修饰电极在含有0.1μg/mL及0.4μg/mL过氧化月桂酰的有机相介质 (溶液体积25mL,乙腈/1,2-二氯乙烷体积比4:1,并含有0.1mol/L硫酸水溶液0.5mL及1.0mol/L氯化锂乙腈溶液1.0mL)中的循环伏安图,结果如图6所示。

图6 HRP酶修饰电极在不同浓度过氧化月桂酰的有机相介质中的循环伏安图(1-0.1μg/mL过氧化月桂酰,2-0.4μg/mL过氧化月桂酰,扫描速度70mV/s)Fig.6 Voltammograms of HRP enzyme modified electrode in organic media including different concentrations of lauroyl peroxide:(1)0.1 μg/mL,(2)0.4 μg/mL,at the scan rate of 70mV/s.
而HRP酶修饰电极,随着过氧化月桂酰加入到有机相介质中,其循环伏安图中的还原峰电流逐渐增加,当过氧化月桂酰浓度从0.1μg/mL上升为0.4μg/mL时,还原峰电流增加了10.52%。而氧化峰逐渐降低,当过氧化月桂酰的浓度为1.2μg/mL时其氧化峰电流就基本消失。这种现象与HRP酶修饰电极的电极过程有关。
当过氧化月桂酰加入到有机相中后,它在HRP酶电极表面快速将HRP(FeⅢ)氧化成氧化态的HRP(FeIV),其自身发生还原反应,而膜内的亚铁氰化钾作为电子介体,能够在HRP酶与电极表面之间传递电子,使氧化态的HRP(FeIV)快速还原,而亚铁氰化钾被氧化,形成的铁氰化钾在电极表面得电子快速被还原,所以当溶液中加入过氧化物时,电极表面的铁氰化钾浓度增加,因此产生的还原峰电流增加,而氧化电流下降,此外由于反应的活化能下降,使得还原峰电位发生负移,其氧化还原过程为:
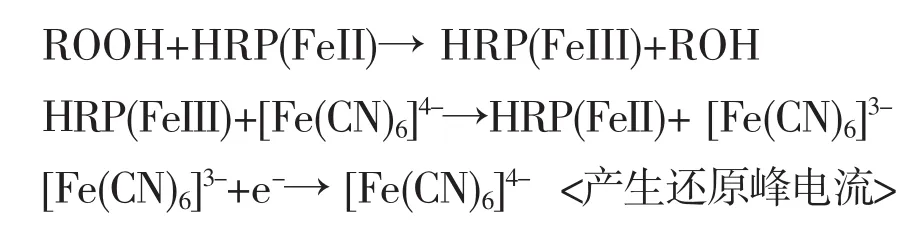
3.3.3 HRP酶修饰电极的电化学反应及电极过程
为了探明HRP酶修饰电极在过氧化物存在时的电极过程,本研究分别记录了0.1μg/mL过氧化月桂酰的溶液中,HRP酶修饰电极在不同电位扫描速度 10、20、30、50、100、150、200mV/s 下的循环伏安曲线。
由结果可知,随着扫描速度的增加,还原峰电流逐渐降低,当扫描速度达到50mV/s时,还原峰电流值为最低,之后随着扫描速度的增加,还原峰电流逐渐增大。分别作还原电流Ipred与扫描速度及扫描速度平方根的关系图,发现扫描速度在10~50mV/s内,Ipred与扫描速度的平方根成线性关系,即 I=-0.1295v-1/2+1.5825(r=0.9945),而扫描速度在50~200mV/s时,Ipred与扫描速度成线性关系,即I=0.003v+0.8584(r=0.9966)。
电极过程一般分为传质过程控制和表面过程控制,当峰电流响应与扫描速度的平方根成线性关系时,可以认定电极过程为传质过程,即反应物运动到电极表面的过程;当峰电流响应与扫描速度成线性关系时,说明电极过程为表面过程控制,即电子转移到电极上的过程[14]。
从本研究结果来看,HRP酶修饰电极在扫描速度较低(<50mV/s)时,以传质过程控制为主,是通过HRP酶传递到电极表面发生还原反应产生还原电流的。随着扫描速度提高,电极的传质过程控制转弱,逐渐转为表面过程控制,此时HRP酶电极的氧化还原反应与电子介体——亚铁氰化钾在电极表面的氧化还原有关,即在溶胶-凝胶修饰膜内亚铁氰化钾以还原态形式存在,在电极表面失电子被氧化,氧化态铁氰化钾能及时在电极表面得电子被还原,由此还原峰电流增大。
根据前人的文献资料,HRP酶修饰电极的电化学反应属于表面电化学控制[15],并没有出现传质过程控制的阶段,推测这种结果或与修饰电极为双层膜结构有关,以往文献中采用的HRP酶电极多为单层膜结构。
3.4 HRP酶修饰电极测定过氧化物电化学参数的选择
由于在有机相介质中的HRP酶修饰电极为完全不可逆体系,因此其峰电位时常会发生位移,为排除电位移动对电流测定的影响,本研究选择计时电流法在恒电位作用下测定HRP酶电极对过氧化物的催化电流。图7为施加电位与HRP酶电极对过氧化月桂酰催化产生的最大还原电流间的关系曲线。

图7 施加电位与HRP酶修饰电极对过氧化月桂酰产生的催化还原电流间的关系Fig7 The influence between the current of the catalytic reduction produced by the applied potential and HRP enzyme electrode to lauroyl peroxide
由图可知,当施加电位较小(0V)时,HRP酶电极产生的还原电流接近0,随着施加电位向负方向移动,催化还原电流逐渐增加,当施加电位达到-0.6V时,电流值达到最大,之后还原电流基本保持稳定,因此本研究选择-0.6V为计时电流法的施加电位。
3.5 HRP酶修饰电极的电流响应与过氧化月桂酰浓度间的关系
采用外标法,配制一系列不同浓度的过氧化月桂酰溶液样品,在-0.6V电位下记录计时电流曲线。每一浓度样品平行测定三次,记录其还原电流并取平均值,之后扣除HRP酶修饰电极在空白溶液中的还原电流,以此作为HRP酶修饰电极对该浓度过氧化月桂酰的催化还原电流响应△Ipred。以过氧化月桂酰浓度为横坐标,△Ipred为纵坐标,绘制线性回归曲线 (图8),由图可知,△Ipred与过氧化月桂酰浓度在0.08~6.4μg/mL范围内具有良好的线性相关关系(R=0.9925),线性回归方程如公式1所述。
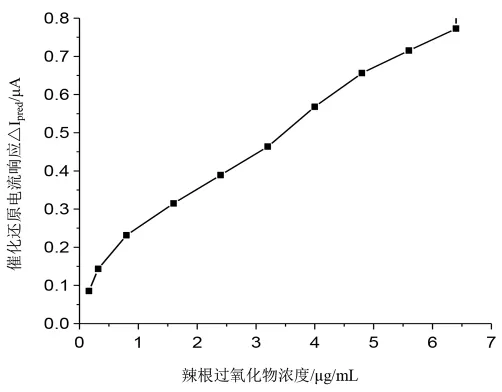
图8 HRP酶修饰电极电流响应与过氧化月桂酰浓度间的校正曲线Fig.8 The calibration curve of the peak current of HRP enzyme electrode to the concentration of lauroyl peroxide

其中,△Ipred为HRP酶电极对过氧化月桂酰的催化还原电流响应△Ipred,μA;C为过氧化物浓度,μg/mL。
3.6 HRP酶电极动力学参数(米氏常数)的测定
表观米氏常数可以表示酶和底物之间的亲和能力,只与酶的性质、底物种类及反应条件有关,与酶的浓度无关[16]。电化学方法测量米氏常数,可通过双倒数作图法获得,遵守Lineweaver-Burk方程[17],如公式2所述。

其中iss为稳态电流,imax为极限电流,C为过氧化物浓度,Km为表观米氏常数。
利用加入不同浓度过氧化月桂酰得到的电极工作曲线,对C和i分别求倒数,得到1/C和1/i,并可直接进行线性拟合,得到y=ax+b,1/imax就是公式中的b,Km/imax为a,利用b和a就可以求得 Km。
在本研究中,根据3.5的校准曲线,以1/iss对1/C作图,其中截距为1.560,斜率为1.666,由此计算得到Km=1.07mmol/L,这比采用交联法[15]制得的HRP酶修饰电极的米氏常数(4.943 mmol/L)小,说明采用溶胶-凝胶法固定的HRP酶能很好的保持其催化活性,使其对过氧化月桂酰具有较大的亲和性。
3.7 HRP酶修饰电极的重现性及使用寿命
为了检验HRP酶电极的重现性,在相同的实验条件下,制备了15支HRP酶电极,取其中的5支酶电极对0.32μg/mL的过氧化月桂酰进行催化还原实验,重复测定5次,计算其平均电流响应值为11.53±0.783μA,相对标准偏差6.8%(n=5)。
另10支HRP酶电极,每5支为一组,于4℃冰箱中保存,分别于存放15天及30天后对0.32μg/mL的过氧化月桂酰溶液进行电化学分析,结果,测定得到的电流响应分别为10.95±0.577μA(存放 15 日,n=5,RSD=5.27%)及 10.67±0.804μA(存放 30 日,n=5,RSD=7.53%)电流响应分别下降了5.03%及7.46%。说明采用二氧化硅的凝胶膜能够较好的保持酶的活性。
3.8 HRP酶电极在实际植物油样品过氧化值检测中的应用
本研究采用与3.5中所述相同的计时电流法,进一步对5种市售植物油(大豆油、花生油、调和油、葵花子油、玉米油)共10个样品的过氧化物进行分析。
同时采用国标碘量法 (GB/T 5538-2005)检测这六种植物油的过氧化值,对两种方法的测定结果进行比对,考察采用HRP酶电极法检测过氧化值的有效性,结果如表2所示。

表2 HRP酶修饰电极法与碘量法的结果比对Table 2 Determination of POV of vegetable oils by the developed method and the Iodometric method
以参考方法的值为横坐标(国标法),新方法的值为纵坐标(HRP酶电极法),绘制曲线并计算曲线的截距、斜率和相关系数,结果如图9所示。
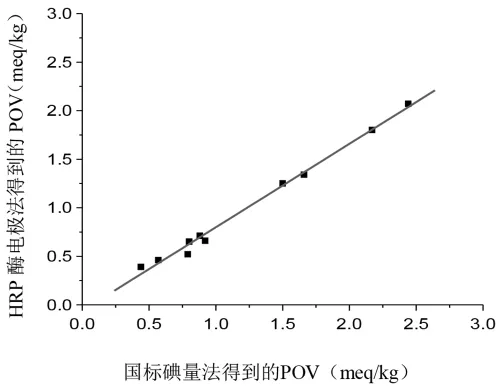
图9 HRP酶电极法准确性评价Fig.9 The accuracy assessment of HRP enzyme electrode method
通常认为,如果两种方法检测的结果完全一致,其回归方程截距应为零,斜率和相关系数应为1,实际上这种情况是不存在的。当相关系数很高,说明新方法的精确性很好,可以用该方法进行定量分析,但并不能说明该方法检测的值与参考方法是一致的。从本研究的结果来看,国标法与HRP酶电极法间建立的回归方程相关系数较高,(Y=1.153X+0.080,R=0.9960) 这说明新方法的精确性很好,但从实际检测结果来看,10种植物油样品采用HRP酶电极法测定得到的过氧化值,全部小于国标法测定的结果,有8种植物油样品|相对误差|在10%~20%之间,有两种样品|相对误差|>20%。两种方法在一致性方面较差,分析原因,可能是由于植物油样品加入到有机相中后,溶液的黏度增大,导致电极表面电子的传递速度进一步下降,从而使HRP酶电极在检测植物油样品时产生的催化还原电流响应比真实值小,相应的过氧化值也比真实值 (比对方法)要小。
4 结论
本研究使用溶胶-凝胶法构建辣根过氧化物(HRP)酶修饰电极,研究了HRP酶电极的催化反应机理及电极过程,优选出制备酶电极的最佳工艺条件,确定了过氧化物浓度与HRP酶电极产生的催还还原电流响应间的相关关系,并将制备得到的电极应用于食用植物油样品的实际检测中,得到如下结论:
采用溶胶-凝胶法构建的双层膜结构HRP酶电极,在有机相介质中能够对过氧化月桂酰产生显著地还原电流响应,作为电子介体的亚铁氰化钾能够有效的在HRP酶与电极表面传递电子,增强还原电流响应;随着过氧化月桂酰添加量的增加,HRP酶电极产生的还原电流逐渐增强,氧化电流逐渐减弱,当过氧化月桂酰浓度达到1.2μg/mL时,氧化峰电流基本消失;通过对还原峰电流与扫描速度间的关系研究,证明当扫描速度较低时HRP酶电极以传质过程控制为主,即通过HRP酶传递到电极表面发生还原反应产生还原电流。当扫描速度较高时,电极的传质过程控制转弱,逐渐转为表面过程控制,此时HRP酶电极的氧化还原反应与电子介体——亚铁氰化钾在电极表面的氧化还原有关;HRP酶修饰电极最佳的制备工艺为正硅酸乙酯、水和甲醇的体积比为1.5∶1.5∶4,并加入一定量的CTAB以改善成膜效果,亚铁氰化钾浓度10mmol/L,HRP酶浓度10mg/mL;采用计时电流法根据不同浓度的过氧化月桂酰在HRP酶电极上的催化还原电流,建立其与过氧化物浓度间的校正曲线,并根据Lineweaver-Burk方程,确定该体系下HRP酶的表观米氏常数Km;采用溶胶-凝胶法构建HRP酶修饰电极,具有一定的重现性与精确度,但在实际植物油样品过氧化值检测中与国标碘量法测定的结果一致性较弱,整体值偏小,分析原因可能是由于植物油的加入使得有机相的黏度上升,还原电流响应比真实值小造成的。
