高同型半胱氨酸血症的诊断、治疗与预防专家共识*
2022-06-07李东晓张宏武王晓建杨艳玲
李东晓 张 尧 张宏武 王 琳 王晓建 杨艳玲
中国妇幼保健协会出生缺陷防治与分子遗传分会、儿童早期发展专委会、儿童疾病和保健分会遗传代谢学组;北京医学会罕见病分会;
中国医师协会儿科分会内分泌遗传代谢学组、青春期医学专业委员会临床遗传学组及生化学组;中华预防医学会残疾预防与控制专业委员会、儿童保健分会;
深圳罕见病代谢组学精准医学工程研究中心;《罕少疾病杂志》编辑委员会
高同型半胱氨酸血症是一种常见的代谢异常,病因复杂,包括遗传性和非遗传性两大类疾病,可自胎儿到老年发病,人群总体患病率高达5%[1]。遗传性高同型半胱氨酸血症已被我国《第一批罕见病目录》收录,是可治疗的遗传代谢病。根据血中总同型半胱氨酸浓度,高同型半胱氨酸血症分为轻型(15~30μmol/L)、中间型(31~100μmol/L)和重型(>100μmol/L)[2]。持续高同型半胱氨酸血症可导致神经精神、心脑血管、肾脏、眼、骨骼等多系统损害,临床表现缺乏特异性,致残、致死率很高[3-5]。采用液相串联质谱法对新生儿进行筛查,可早期发现高同型半胱氨酸血症1型及甲基丙二酸血症合并同型半胱氨酸血症等部分患者[6-8]。其他原因所致高同型半胱氨酸血症患者需通过临床调查、生化代谢检查、基因检测等综合分析才能明确病因[9]。由于患者病因各异、临床表型轻重不同,需要个体化治疗。如果能早期诊治,绝大部分患者预后良好。近年来高同型半胱氨酸血症领域取得了许多进展,诊断及治疗策略不断更新。国内专家在不同领域发表了相关共识[4]。
为进一步规范高同型半胱氨酸血症的诊断与治疗,中国妇幼保健协会(出生缺陷防治与分子遗传分会、儿童早期发展专委会、儿童疾病和保健分会遗传代谢学组)、北京医学会罕见病分会、中国医师协会(儿科分会内分泌遗传代谢学组、青春期医学专业委员会临床遗传学组及生化学组)、中华预防医学会(残疾预防与控制专业委员会、儿童保健分会)、深圳罕见病代谢组学精准医学工程研究中心及《罕少疾病杂志》编辑部就高同型半胱氨酸血症的诊断、治疗和防控策略进行讨论,并参考国内外的经验及指南,达成以下共识。
1 病因及分类
同型半胱氨酸是蛋氨酸代谢过程中的中间产物,主要通过甲基化及转硫化两条途径转化代谢,血液同型半胱氨酸水平受参与蛋氨酸代谢过程中的各种酶及辅因子(如钴胺素、维生素B6、叶酸)影响[10]。食物中的叶酸还原为四氢叶酸,在丝氨酸羟甲基转移酶作用下转化为5,10-亚甲基四氢叶酸,并在亚甲基四氢叶酸还原酶 (methylenetetrahydrofolate reductase,MTHFR)作用下转化为5-甲基四氢叶酸,提供甲基给同型半胱氨酸,在蛋氨酸合成酶作用下生成蛋氨酸。钴胺素参与5-甲基四氢叶酸的再甲基化反应。蛋氨酸在蛋氨酸腺苷转移酶作用下,被三磷酸腺苷激活形成S-腺苷蛋氨酸,S-腺苷蛋氨酸是体内最重要的甲基供体。在转硫途径中,同型半胱氨酸在胱硫醚β-合成酶作用下转化为胱硫醚,并进一步在胱硫醚γ-裂解酶作用下裂解为半胱氨酸和α-酮丁酸[10]。在同型半胱氨酸再甲基化及转硫通路中,任何一个酶或辅酶缺陷都会导致高同型半胱氨酸血症(表1)。
遗传性高同型半胱氨酸血症分为酶缺陷和辅酶缺陷两大类,酶缺陷以胱硫醚β-合成酶、亚甲基四氢叶酸还原酶、蛋氨酸腺苷转移酶缺陷最为常见,辅酶缺陷以钴胺素和叶酸代谢障碍最为常见(表1)[10-11]。
一些非遗传性因素也可导致高同型半胱氨酸血症,如营养不良、长期素食导致的营养素缺乏(如叶酸、维生素B6、钴胺素、甜菜碱缺乏)、衰老、慢性胃肠疾病、肝病、肾病、恶性肿瘤、药物(如异烟肼、甲氨蝶呤)以及不良生活方式(如长期吸烟、酗酒等)(表1)[4,12-13]。
2 诊断与鉴别诊断
不同病因导致的高同型半胱氨酸血症患者体内生化代谢异常有所差异,需通过病史调查、血液及尿液代谢分析、维生素测定、基因分析等检查鉴别诊断(表1)。
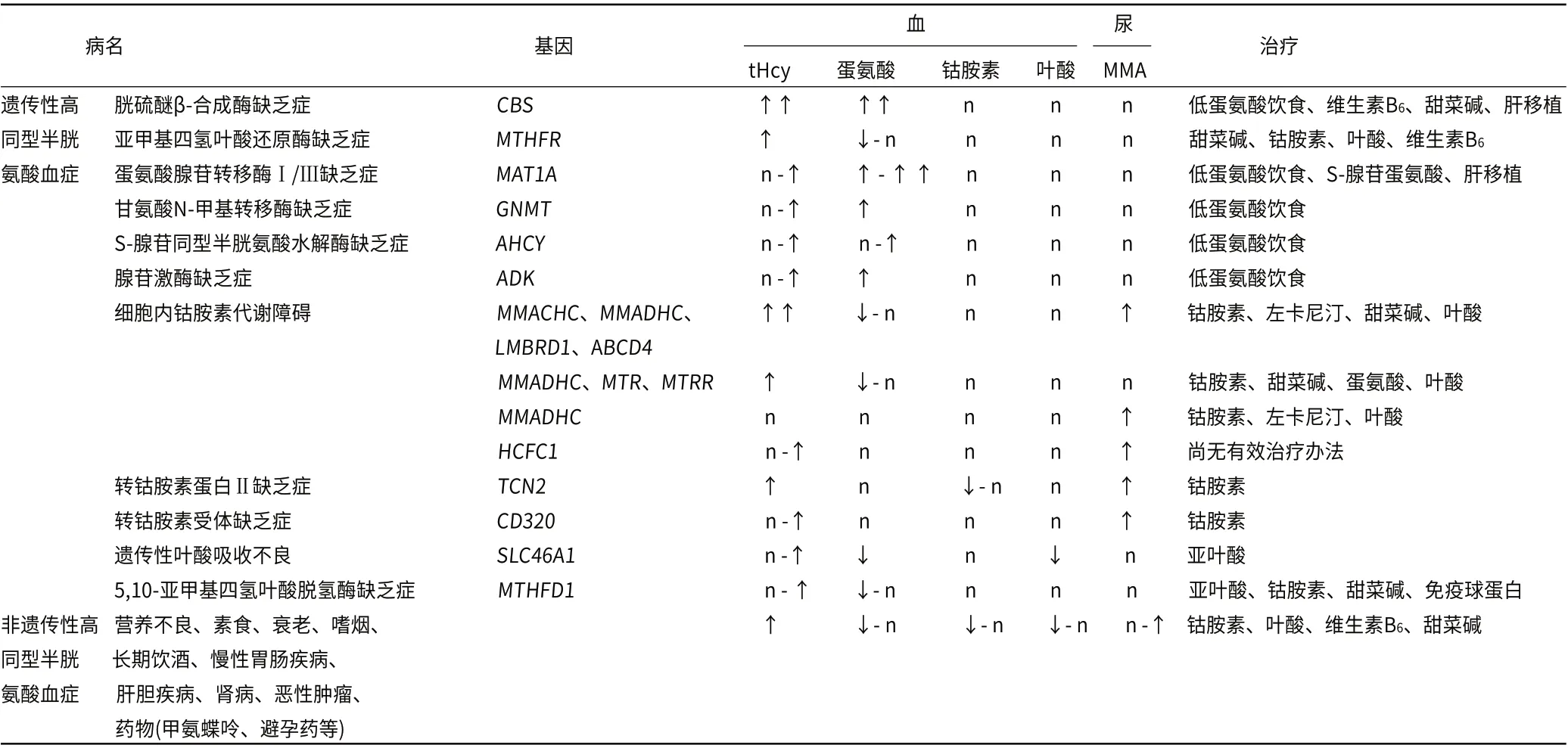
表1 高同型半胱氨酸血症的病因、生化特点及治疗
(1)病史调查:一般资料、症状、体征,调查个人史、饮食习惯、用药史、家族史。对于哺乳期婴儿,需调查母亲的饮食与营养状况及疾病史,以鉴别母亲营养不良导致的婴儿继发性高同型半胱氨酸血症[4,12]。
(2)血浆或血清总同型半胱氨酸:血液中大部分同型半胱氨酸与蛋白质结合,少数与同型半胱氨酸自身或半胱氨酸结合形成混合型同型半胱氨酸二硫化物,推荐检测总同型半胱氨酸浓度作为确诊高同型半胱氨酸血症的关键方法[14-15]。
(3)血液维生素测定:检测血清叶酸、钴胺素、维生素B6水平,鉴别是否营养因素缺乏导致的继发性高同型半胱氨酸血症[16-17]。
(4)血液氨基酸、游离肉碱及酰基肉碱谱:采用串联质谱法,可确诊一些疾病,如同型半胱氨酸血症1型及高蛋氨酸血症患者蛋氨酸显著增高[6,18],甲基丙二酸血症合并同型半胱氨酸血症患者丙酰肉碱增高,蛋氨酸正常或降低[7,19-20],同型半胱氨酸血症2型患者丙酰肉碱正常,蛋氨酸正常或降低[21-22]。
(5)尿有机酸:需采用气相色谱质谱法,甲基丙二酸血症合并同型半胱氨酸血症患者尿液甲基丙二酸显著增高[7,19,23],高同型半胱氨酸血症1型及高蛋氨酸血症患者尿有机酸正常[8,18]。
(6)脏器功能评估:如肝肾功能、眼部、心电图、超声心动图、脑CT及MRI、精神行为评估等,了解患者脏器受损的情况[1,8]。
(7)基因分析:可采用高通量测序或Sanger测序等技术,明确遗传性高同型半胱氨酸血症的基因缺陷。
3 遗传缺陷所致高同型半胱氨酸血症的诊断与治疗
已知多种遗传病可引起高同型半胱氨酸血症,患者病情轻重不同,症状、体征及生化代谢异常交叉重叠,需要明确病因及脏器功能,个体化精准治疗(表1)。
3.1 胱硫醚β-合成酶(cystathionine beta-synthase,CBS)缺乏症(OMIM 236200)又称高同型半胱氨酸血症1型或经典型同型半胱氨酸尿症,病因为CBS基因(OMIM 613381)缺陷,为常染色体隐性遗传病,国内研究较少,美国报道发病率约为1/200 000[2]。胱硫醚β-合成酶是转硫化通路中的第一个限速酶,维生素B6为其辅酶,通过腺苷蛋氨酸的变构激活参与转硫途径。患者多于学龄前~青少年期发病,主要表现为智力运动落后或倒退、近视、晶状体脱位、马凡综合征体型、血栓及心脑血管病[8,24],患者血、尿总同型半胱氨酸和蛋氨酸显著升高,可通过新生儿筛查检出高蛋氨酸血症被发现,通过CBS基因分析确诊(表1)[8,20,25]。
根据患者对维生素B6治疗的反应,高同型半胱氨酸血症1型分为轻型的维生素B6反应型和较严重的维生素B6无反应型。为判断维生素B6的有效性,患者可连续服用2~6周维生素B6100mg/日~500mg/日,检测用药前后的血总同型半胱氨酸,若治疗后血总同型半胱氨酸<50μmol/L,为维生素B6反应型;若降幅超过20%,考虑为维生素B6部分反应型;若降幅低于20%,则为维生素B6无反应型[25-26]。
治疗目标是血总同型半胱氨酸<50μmol/L。推荐采用最小剂量维生素B6,一般不大于10mg/(kg·日),总量<500mg/日,以减少外周神经炎等副作用。对于维生素B6部分反应型或无反应型患者可给予维生素B650~100mg/日,同时需限制蛋氨酸摄入,给予不含蛋氨酸的特殊医学配方粉,根据血总同型半胱氨酸和蛋氨酸水平动态调整饮食。甜菜碱有助于改善蛋氨酸代谢,儿童150~200mg/(kg·日),成人6~9g/日,每日分2~3次口服[25]。为保证营养,可给予叶酸(1~5mg/日)、钴胺素等维生素。对于维生素B6无效型患者,可选择肝移植治疗[27]。
3.2 亚甲基四氢叶酸还原酶(methylenetetrahydrofolatereductase,MTHFR)缺乏症 MTHFR缺乏症又称高同型半胱氨酸血症2型(OMIM 236250),是一种常染色体隐性遗传病,由于MTHFR基因变异所致,是高同型半胱氨酸血症最常见的遗传学病因。MTHFR是一碳单位代谢过程中叶酸代谢的关键酶,MTHFR基因缺陷使MTHFR酶功能丧失或活性减低,导致甲基化受损及血同型半胱氨酸升高。MTHFR变异对血浆同型半胱氨酸水平的影响有量效关系,单等位基因杂合变异可能会引起血浆同型半胱氨酸轻到中度升高,但纯合变异往往引起严重的高同型半胱氨酸血症[28],应结合患者MTHFR基因型、血浆同型半胱氨酸水平及临床表型综合分析(表1)。
MTHFR缺乏症是引起心脑血管病、神经精神疾病的主要原因之一[22,29],也是导致胎儿神经管畸形、先天性心脏病等出生缺陷的高危因素[13,28,30]。患者临床表型复杂,轻重不同,个体差异显著,可于新生儿至成人各个年龄段发病。重症患者于新生儿期或婴儿早期发病,常表现为喂养困难、智力运动落后、脑积水、癫痫发作。婴儿晚期和儿童期发病的患者多表现为癫痫、认知损害、行为异常、小头畸形[21]。年长儿及成年发病的患者病情更为复杂,表现为智力运动倒退、周围神经病、步态异常,部分患者以精神行为异常、精神分裂症形式起病,一些患者因高血压、心脑血管病、心肌病被发现,严重者死于血栓栓塞[22,31-32]。
MTHFR缺乏症患者的血同型半胱氨酸浓度差异显著,严重患者新生儿期血同型半胱氨酸显著增高,而轻症患者随着病程进展血同型半胱氨酸逐渐升高,同一名患者可以经历血同型半胱氨酸从正常到显著增高的演变。患者血蛋氨酸浓度偏低或正常,少数患者合并叶酸缺乏及巨幼细胞性贫血,确诊需依靠MTHFR基因检测[22,32]。
MTHFR缺乏症的治疗需根据患者个体情况,给予叶酸或亚叶酸(5~30mg/日)、钴胺素(0.5~2mg/日)、维生素B6(30~100mg/日)、甜菜碱(1~6g/日)及蛋氨酸(40~50mg/kg·日),并对症治疗[21,33],多数患者好转或康复。鉴于高同型半胱氨酸血症与高血压、脑卒中风险呈正相关,《中国高血压防治指南2018年修订版》建议高血压伴同型半胱氨酸升高的患者适当食用新鲜蔬菜水果,必要时补充叶酸[5]。
3.3 细胞内钴胺素代谢障碍已知多种遗传缺陷可导致钴胺素相关再甲基化障碍,钴胺素转运及代谢途径中任何酶缺陷均可导致高同型半胱氨酸血症。钴胺素在溶酶体膜蛋白cblF及cblJ作用下变成三价钴胺素释放到细胞质,与cblC结合后修饰加工成二价钴胺素,经cblD蛋白与cblC蛋白作用使二价钴胺素分化形成甲钴胺及腺苷钴胺。甲钴胺作为蛋氨酸合成酶的辅酶参与再甲基化途径;腺苷钴胺作为甲基丙二酰辅酶A变位酶的辅酶参与甲基丙二酰辅酶A向琥珀酰辅酶A的转化[16-17]。cblX可能通过cblC影响钴胺素代谢(表1)[34]。
cblC、cblF、cblJ缺陷同时影响了甲钴胺和腺苷钴胺的形成,生化表型为甲基丙二酸血症和高同型半胱氨酸血症[7,35-36]。cblD缺陷生化表型可以是单纯高同型半胱氨酸血症、单纯甲基丙二酸血症,或甲基丙二酸血症合并同型半胱氨酸血症。cblE、cblG缺陷患者的生化表型为单纯高同型半胱氨酸血症[35,37]。cblC、cblD、cblE、cblF、cblG、cblJ及cblX蛋白分别由MMACHC、MMADHC、MTRR、LMBRD1、MTR、ABCD4及HCFC1基因编码,除cblX为X连锁遗传外,其余均为常染色体隐性遗传(表1)[34-35,37]。
cblC型是甲基丙二酸血症合并同型半胱氨酸血症最常见的类型,国内外在筛查、诊断、治疗及预防方面积累了丰富的经验。患者临床表型差异较大,可自胎儿到成年发病,可导致脑、心、肾、血液、眼等多系统损害,引起智力运动发育落后或倒退、癫痫、脑积水、肺动脉高压、肾功能不全、溶血尿毒综合征、血栓等多种疾病,常合并巨幼细胞性贫血、低蛋氨酸血症,血清叶酸、钴胺素水平正常,血液总同型半胱氨酸及尿甲基丙二酸显著增高[19,38-39]。
cblC型钴胺素治疗有效,急性期静脉注射或肌内注射甲钴胺(1~10mg/日)、或肌内注射腺苷钴胺或羟钴胺(1~10mg/日),静滴或口服左卡尼汀100~300mg/kg·日,静脉补液,纠正代谢性酸中毒,对症治疗,一般无需限制天然蛋白质。病情稳定后,钴胺素长期维持,根据个体情况每次肌注1~10mg,每周1~3次;口服左卡尼汀30~50mg/kg·日,将血液游离肉碱水平维持在30~100μmol/L;口服甜菜碱1~6g/日;合并贫血的患儿可口服叶酸或亚叶酸5~15mg/日;对严重蛋氨酸缺乏的患者,可补充蛋氨酸,对于合并脑积水的患者,必要时手术治疗[7,37,40]。cblX型患者生化表型为尿液甲基丙二酸增高,可伴或不伴高同型半胱氨酸血症,患者血液酰基肉碱谱可能正常,采用传统的液相串联质谱法新生儿筛查可能漏诊,尿有机酸分析及基因分析是诊断的关键[41]。患者病情严重,常表现为重度智力运动障碍、难治性癫痫,伴不同程度的畸形、锥体外系症状等,目前尚无有效的方法,采用针对cblC型的方法无明显效果,预后不良[34,41]。
3.4 钴胺素吸收和转运障碍已知内因子、转钴胺素蛋白Ⅰ(transcobalaminⅠ,TC Ⅰ)、转钴胺素蛋白Ⅱ(transcobalamin Ⅱ,TC Ⅱ)参与钴胺素的吸收和转运,每个蛋白或其受体功能缺陷均可导致钴胺素缺乏。其中TC Ⅱ缺乏症和转钴胺素受体缺乏症常导致甲基丙二酸血症和高同型半胱氨酸血症(表1)[16,42]。
3.4.1 TC Ⅱ缺乏症 TC Ⅱ是钴胺素的主要转运蛋白,由TCN2基因编码。TC Ⅱ缺乏症是一种罕见的常染色体隐性遗传病,可导致胎儿死亡、发育落后、腹泻、巨幼细胞性贫血、全血细胞减少症、神经精神异常以及免疫功能低下等,生化表型为甲基丙二酸血症合并同型半胱氨酸血症。肌内注射或口服钴胺素可有效改善患者病情(表1)[43]。
3.4.2 转钴胺素受体(transcobalamin receptor,TCblR)缺乏症 又称甲基丙二酸血症合并同型半胱氨酸血症TCblR型,是一种罕见的常染色体隐性遗传病,转钴胺受体功能下降导致细胞摄取钴胺素减少,引起甲基丙二酸血症。患者早期通常无症状,血中丙酰肉碱升高,总同型半胱氨酸水平正常到中度升高,少数患者出现视网膜血管闭塞性疾病,视力丧失。肌内注射或口服钴胺素可有效改善患者病情(表1)[44]。
3.5 叶酸吸收和代谢障碍已知遗传性叶酸吸收不良和5,10-亚甲基四氢叶酸脱氢酶(5,10-methylene-tetrahydrofolate dehydrogenase,MTHFD)缺乏症是导致脑叶酸缺乏症及高同型半胱氨酸血症的病因(表1)[42,45-46]。
遗传性叶酸吸收不良是一种罕见的常染色体隐性遗传病,由于SLC46A1基因编码的质子偶联叶酸转运子功能异常导致肠道叶酸吸收障碍,并且叶酸不能通过血脑屏障转运进入脑内。患者常在婴幼儿期发病,表现为巨幼细胞性贫血、生长发育障碍、腹泻、口腔溃疡、智力运动落后、癫痫、行为异常、低丙种球蛋白血症等,部分患者伴颅内钙化。患者脑脊液5-甲基四氢叶酸显著降低,伴或不伴高同型半胱氨酸血症。由于叶酸与叶酸受体结合阻碍叶酸转运,不推荐应用叶酸治疗,口服或肌注亚叶酸有效(表1)[45,47]。
MTHFD缺乏症是一种极罕见的常染色体隐性遗传病,又称联合免疫缺陷合并巨幼细胞性贫血,伴或不伴高同型半胱氨酸血症,是由于MTHFD1基因缺陷导致的先天性叶酸代谢障碍。患者主要临床表现为溶血尿毒综合征、巨红细胞症、癫痫、听力障碍、视网膜病变、轻度智力低下、所有亚群淋巴细胞减少和低T细胞受体切除环,生化表型为高同型半胱氨酸血症、低蛋氨酸血症,血叶酸、钴胺素水平正常[48]。亚叶酸、钴胺素和甜菜碱为有效治疗方法,可显著改善症状和生化异常,但不能纠正视网膜病变。对于免疫功能低下的患者,可补充免疫球蛋白,预防性应用甲氧苄啶和磺胺甲恶唑预防严重感染(表1)[49]。
3.6 原发性高蛋氨酸血症已知多种酶缺陷可导致原发性高蛋氨酸血症,蛋氨酸腺苷转移酶Ⅰ/Ⅲ(methionine adenosyltransferase Ⅰ/Ⅲ deficiency, MATⅠ/Ⅲ) 缺乏症最常见,甘氨酸N-甲基转移酶缺乏症、S-腺苷同型半胱氨酸水解酶缺乏症及腺苷激酶缺乏症罕见。患者血蛋氨酸显著增高,可通过新生儿筛查检出,总同型半胱氨酸浓度大多正常,少部分轻中度增高(表1)[18,50]。
蛋氨酸腺苷转移酶Ⅰ/Ⅲ缺乏症患者在婴幼儿期大多无明显症状,随着病程进展及血液蛋氨酸、总同型半胱氨酸持续增高,少数患者出现脑水肿、脑白质脱髓鞘病变、智力运动发育迟缓或倒退、精神异常、癫痫及体臭等,因此不能认为蛋氨酸腺苷转移酶Ⅰ/Ⅲ缺乏症所致高蛋氨酸血症是一种良性疾病[18,50]。甘氨酸N-甲基转移酶缺乏症极为罕见,迄今仅报道数例,病情较轻,无症状或轻度肝肿大,可伴转氨酶增高[51-52]。S-腺苷同型半胱氨酸水解酶缺乏症患者常有多系统损害,主要表现为发育迟缓、肌病、肝功能损害,一些患者发生胆汁淤积及肝细胞癌等并发症,常于婴儿期后出现高蛋氨酸血症,多伴有血清肌酸激酶增高[53]。腺苷激酶缺乏症则更为严重,主要表现为脑病、全面发育迟缓、肝功能损害、面部畸形(如前额凸出、大头畸形、眼距宽和鼻梁低平)[54]。
关于原发性高蛋氨酸血症的治疗方法目前尚不明确。建议对于血蛋氨酸浓度超过800μmol/L的患者限制蛋氨酸摄入,争取使血蛋氨酸降至500μmol/L以下。对于有症状的患者,蛋氨酸超过600μmol/L时,需限制蛋氨酸摄入,若限制蛋氨酸饮食治疗期间出现症状和/或体征,可补充腺苷蛋氨酸(400~1600 mg/日)[11,18]。低蛋氨酸饮食可能对部分患者有益,如果患者血液蛋氨酸持续高于800μmol/L,应考虑肝移植治疗[55]。
4 非遗传缺陷所致获得性高同型半胱氨酸血症的治疗
多种非遗传性疾病可导致高同型半胱氨酸血症,例如营养不良、素食、偏食、衰老、嗜烟、长期饮酒、慢性胃肠疾病、肝胆疾病、肾病、恶性肿瘤、药物(甲氨蝶呤、避孕药等),引起获得性叶酸、钴胺素、维生素B6或甜菜碱缺乏,需在针对病因治疗的基础上,均衡饮食,补充叶酸、钴胺素、维生素B6或甜菜碱,改善营养代谢状况[4,12,16]。
5 遗传咨询和产前诊断
高同型半胱氨酸血症不仅损害患者多脏器功能,也会影响精子、卵子及胚胎的发育,导致胎儿先天畸形或功能缺陷,通过三级防控可有效降低本病导致的伤残及出生缺陷。
(1)新生儿筛查及高危筛查:采用液相串联质谱法检测血液氨基酸及肉碱谱,可以检出高同型半胱氨酸血症1型、甲基丙二酸血症合并同型半胱氨酸血症及高蛋氨酸血症等疾病[6,19],血清或血浆总同型半胱氨酸测定、维生素检测、尿有机酸分析可进一步鉴别病因[9,23]。
(2)家族成员的风险及携带者筛查:绝大多数高同型半胱氨酸血症为常染色体隐性遗传病,患者的父母携带单个基因杂合变异,多无临床症状。患者的同胞有1/4的几率患病,1/2的可能性为杂合变异的携带者,1/4的可能性不携带致病变异。因此,患者的父母、同胞等家族成员应进行基因筛查及血液总同型半胱氨酸测定,发现携带者及症状早期的患者,应早期干预。补充叶酸、钴胺素等维生素有助于提高生殖质量,预防出生缺陷[13,56]。
成年患者在准备生育之前,需检测配偶的基因,若配偶未携带相同基因的致病变异,则后代患高同型半胱氨酸血症的风险较低。若配偶为致病基因变异携带者,则后代有50%可能为高同型半胱氨酸血症患者,应进行产前诊断。
(3)产前诊断:遗传性高同型半胱氨酸血症产前诊断的必要条件是家族中的先证者基因诊断明确。通常在母亲妊娠9~14周采取胎盘绒毛,或于妊娠16~22周通过羊膜腔穿刺获取羊水,检测胎儿基因。胚胎植入前诊断将辅助生殖与遗传诊断相结合,通过对植入前胚胎的基因分析,挑选正常的胚胎移植,可阻止家族中遗传性高同型半胱氨酸血症再发[57-58]。
羊水同型半胱氨酸检测与质谱技术联合应用有助于cblC缺陷的产前诊断,若胎儿为cblC缺陷,羊水中总同型半胱氨酸、甲基丙二酸、丙酰肉碱增高。该方法方便快捷,有助于弥补先证者临床诊断但基因诊断不明的家庭胎儿的产前诊断,但同时需注意羊水甲基丙二酸、甲基枸橼酸检测存在假阴性的可能[57-58]。
6 小 结
同型半胱氨酸是国内外公认的心脑血管疾病、神经精神疾病及出生缺陷的风险标志物,血液总同型半胱氨酸、血氨基酸及酰基肉碱谱、尿甲基丙二酸检测是高同型半胱氨酸血症生化诊断的关键,临床调查、血液叶酸及钴胺素测定、基因检测是病因诊断的关键。高同型半胱氨酸血症是一类可治疗的代谢病,通过饮食、药物等综合干预,绝大多数预后较好。对于基因诊断明确的患者及其家族成员,通过遗传咨询及产前诊断等干预,可减少家族疾病再发的风险。
执笔者:李东晓(河南省儿童医院);张尧(北京大学第一医院);张宏武(北京大学第一医院);王琳(首都儿科研究所附属儿童医院);王晓建(中国医学科学院阜外医院);杨艳玲(北京大学第一医院)。
专家组成员:(按单位及姓名汉语拼音顺序排序):北京大学第一医院(董慧,窦攀,刘雪芹,秦伦,王朝霞,熊晖,姚红新);北京大学第三医院(闫丽莹);北京协和医院(杜函泽,荆志成);重庆医科大学附属儿童医院(郭艺,朱岷);复旦大学附属儿科医院(陆炜,罗飞宏);广州市妇女儿童医疗中心(黄永兰,刘丽);河北医科大学第二医院(张会丰);河南省儿童医院(陈永兴,王海军,卫海燕);河南省人民医院(韩雄);河南中医药大学第一附属医院(刘新灿,马丙祥,郑宏);湖南省儿童医院(王华);深圳罕见病代谢组学精准医学工程研究中心(姜盼盼,吴莉萍,杨江涛,杨旭);华中科技大学同济医学院附属同济医院(罗小平);华中科技大学同济医学院附属武汉儿童医院(陈晓红);吉林大学白求恩第一医院(杜红伟,梁建民,吕国悦,张一宁);江西省儿童医院(杨玉);赣州市妇幼保健院(刘诗贤,张峰);山东省济南市妇幼保健院(韩炳娟,邹卉);山东省立医院(商晓红);山西省儿童医院(关函洲,阎亚琼,张改秀);山西医科大学第一医院(武师润);上海交通大学医学院附属新华医院(邱文娟,韩连书,顾学范,余永国);上海交通大学医学院附属上海儿童医学中心(王剑);上海交通大学附属儿童医院(李嫔);首都儿科研究所(张彤);首都医科大学附属北京儿童医院(巩纯秀);首都医科大学附属北京妇产医院(孔元原,庄太凤);首都医科大学附属北京友谊医院(孙丽莹,朱志军);首都医科大学附属北京安贞医院(顾虹);四川省成都市第一人民医院(董小丽);福建省厦门大学附属第一医院(刘登礼);厦门大学附属妇女儿童医院、厦门市妇幼保健院(陆妹);郑州大学第三附属医院(贾天明,赵德华,朱登纳);郑州大学第一附属医院(吴静);浙江大学医学院附属第二医院(冯建华);浙江大学医学院附属儿童医院(黄新文,杨茹莱);中国科学院大学深圳医院(吴本清);中国人民解放军总医院第一医学中心(邹丽萍);中国人民解放军总医院第五医学中心(朱世殊);中国医科大学附属盛京医院(麻宏伟,王华);中日友好医院(李晓雯,张知新);中国医学科学院阜外医院(吴艳)。
