基于ADH3基因的野生大麦系统进化分析
2022-06-06王梦薇周羽王树杰沈辉任喜峰
王梦薇,周羽,王树杰,沈辉,任喜峰
1.华中农业大学植物科学技术学院,武汉 430070;2.河南省驻马店市农业科学院,驻马店 463000;3.湖北省襄阳市原种繁殖场,襄阳 441004
大麦是世界上最早被驯化的作物[1]。因其具有早熟、丰产、抗逆性强和适应性广等优点,成为世界广泛栽培的作物之一[2]。野生大麦作为栽培大麦的祖先,其在长期自然选择过程中,积累了大量抗病虫害、抗旱、耐盐、耐贫瘠等优良基因[3],是大麦进行遗传育种和品种改良的重要基因资源[2]。然而,野生大麦的系统演化机制尚存在很大的分歧,一些研究者认为近东新月沃地是大麦的唯一起源地[1,4-9],也有学者支持近东、中国西藏、中亚、埃及、利比亚和埃塞俄比亚等大麦多起源理论[10-15]。目前,大量的分子证据证实,除了近东新月沃地,中亚和中国西藏是大麦的潜在驯化中心[12,15-16]。也有学者证实西藏是大麦的多样性中心而非起源中心,因为找不到野生群落的存在,西藏野生大麦实际上是以杂草的形式存在于田埂的半野生大麦[6]。另据报道,西藏的大麦是近东野生大麦向东扩散到中亚后,继续向东由巴基斯坦、印度扩散到中国西藏[1,6],也有学者认为西藏大麦是由近东通过青藏高原以北地区进入东亚,再到达东南部青藏高原[17-18]。
随着分子生物学的发展,利用基因和基因组重测序等分子手段可更高效地研究大麦的进化及驯化,乙醇脱氢酶基因(ADH)具有催化乙醇分解的生物学功能,在禾本科植物遗传研究中被广泛应用,研究表明该基因在大麦中含有3 个拷贝[19];乙醇脱氢酶Ⅲ基因(ADH3)可揭示大麦种群的地理分布格局,也可用于检测大麦属植物在进化中曾发生的重组和渐渗事件[20]。本研究基于ADH3基因对91份来自近东、中亚和中国西藏的野生大麦进行系统进化分析,明确不同地理区域野生大麦的遗传进化关系,旨在为研究栽培大麦的起源提供参考,为野生大麦的优异种质资源的收集和利用提供依据。
1 材料与方法
1.1 试验材料
本试验材料为91份野生大麦,按地理来源分为3个群体,其中近东野生大麦48份,西藏野生大麦20份,中亚野生大麦23份(表1)。这些野生大麦种质资源均由华中农业大学麦作课题组提供。所有材料种植于华中农业大学校内试验田,每份材料1 行,行长1.5 m,行距20 cm。每份材料在3 叶期取10 株叶片混样提取DNA。

表1 91份野生大麦种质信息Table 1 91 germplasm information of wild barley
1.2 DNA扩增和测序
乙醇脱氢酶Ⅲ基因(ADH3)的引物设计和PCR扩增均按Lin 等[19]的方法,该基因扩增片段为810 bp,PCR 产物经QIAquickTM PCR 纯化试剂盒(Qiagen 公司)进行纯化后,送泰和生物技术有限公司进行Sanger 测序。为了提高测序数据质量,每个样本独立扩增3 次并测序,且正向和反向链均独立测序,分析序列长度为736 bp。
1.3 数据分析
用软件MAFFT 对序列进行比对,将比对后的nex 格式文件导入 PAUP4 中,再从 Modeltest 软件中选56个模型放入PAUP4中搜索最佳模型,根据最佳模型运用MrBayes 软件构建系统发育树。一直增加‘mcmc ngen’的值,使其达到30 000 000,此时的‘average standard deviation of split frequencies’小 于0.01,即已构建出能收敛的系统发育树。之后将‘sump burnin’与‘sumt burnin=’均设置为500,构建出的共识树最后利用Treeview 软件进行可视化;使用STRUCTURE v.2.3.4软件进行群体结构分析;在混合模型下,首先对K从2到4分别进行10次独立模拟运算,每次模拟进行500 000 次预迭代(length of burn-in period)和1 000 000 次基于马尔可夫链蒙特卡罗迭代(Markov Chain Monte Carlo,MCMC)。之后利用Structure Harvester与Clumpak对群体结构的结果计算后验概率值(LnP(D))以及K为2~4 时计算每份种质的遗传成分。对比后的序列用DnaSP6软件进行单倍型分析,最后在PopART 软件中制作单倍型网络图。
2 结果与分析
2.1 野生大麦ADH3基因序列的遗传分析
将本研究的91份不同来源野生大麦材料基于ADH3基因序列的多态性位点数(S)、单倍型个数(H)、单倍型多样性(Hd)、核苷酸多样性(π)以及沃特森估计值(θW)等指标进行遗传差异分析(表2)。结果显示,3 个不同来源野生大麦群体的核苷酸多态性突变位点总数为48 个,其中近东野生大麦具有最多的多态性位点个数(S=46),几乎涵盖了所有多态性位点数;而中亚与西藏野生大麦的多态性位点数量较少,分别为35 个和31 个。从单倍型个数看,3 个野生大麦群体总的单倍型个数为26,近东野生大麦的单倍型个数最多(H=22),中亚与西藏野生大麦的单倍型个数分别为8 和5。从单倍型多样性看,近东野生大麦群体的单倍型多样性最高,为0.914;而中亚与西藏野生大麦的单倍型多样性较低,分别为0.810 和0.753。从核苷酸多样性看,近东野生大麦群体的π值最高,为0.012 65±0.002 47;其次是西藏野生大麦,为0.012 16±0.004 38;中亚野生大麦最低,为0.011 10±0.004 19。从沃特森估计值结果看,近东野生大麦θW值最高,为0.014 40;而西藏野生大麦的θW值最低,为0.012 14。综上,近东野生大麦的多态性位点数、单倍型个数及遗传多样性均远高于中亚和西藏野生大麦。

表2 野生大麦ADH3基因的遗传多样性Table 2 Genetic diversity of ADH3 gene in wild barley
2.2 野生大麦ADH3基因序列的系统进化分析
为了研究3 个不同来源野生大麦群体单倍型之间的关联性,对26 个不同的单倍型进行关联分析(图1)。结果显示,3 个群体的不同单倍型以近东和西藏野生大麦共有的单倍型Hap9 为一个大的多样性中心点,向周围进行点状发散,推断Hap9 可能是一个古老单倍型。其次是近东和中亚野生大麦共有的单倍型Hap1 和3 个地区野生大麦共有的单倍型Hap10也各自形成一个小的多样性中心。此外,近东野生大麦存在特有的单倍型共15 个,而中亚和西藏野生大麦特有的单倍型分别只有2 个(Hap3 和Hap5)和1 个(Hap24)。这更进一步奠定了近东作为大麦起源中心的地位,同时,由于中亚和西藏独特的地理环境增加了新的单倍型出现。另外,近东野生大麦与西藏野生大麦群体的单倍型相距较近,推测西藏野生大麦可能来源于近东野生大麦。
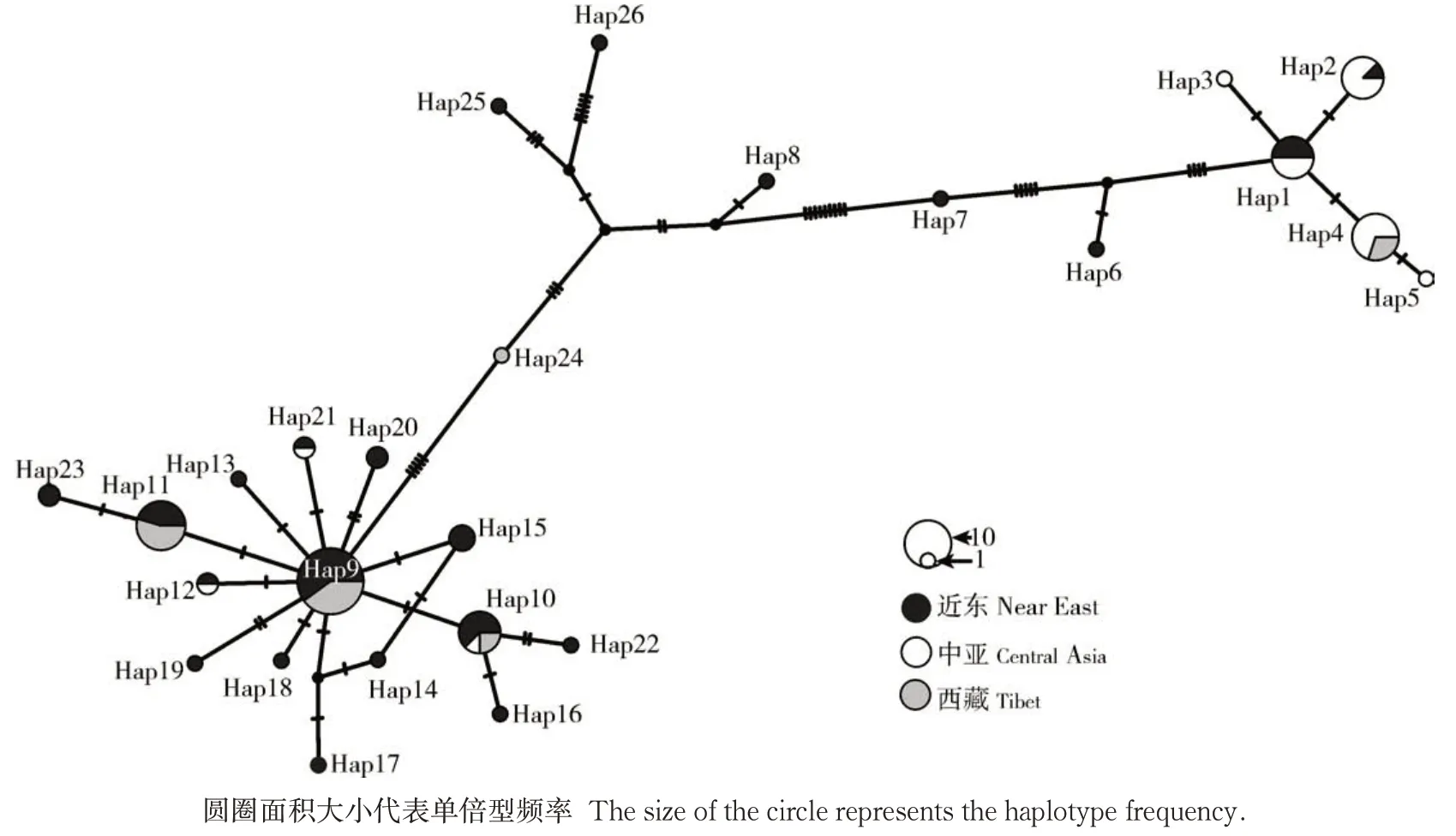
图1 野生大麦ADH3基因序列的单倍型网络Fig.1 Haplotype network of ADH3 gene sequences in wild barley
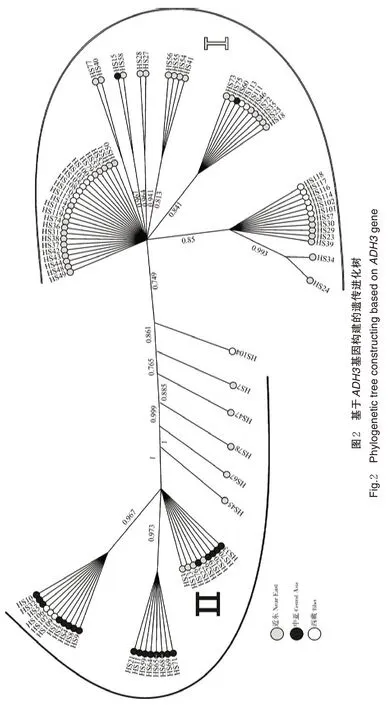
为进一步探究3 个不同野生大麦群体之间的遗传进化关系,基于ADH3基因序列对91份不同来源野生大麦材料构建遗传进化树(图2)。结果显示,91份不同来源野生大麦材料主要被分成2 个大的类群(Ⅰ和Ⅱ),每个类群又分成3~4 个亚群。且81%的近东野生大麦和80%的西藏野生大麦聚类于Ⅰ类群,91%的中亚野生大麦与少部分近东和西藏野生大麦混合聚类于Ⅱ类群,从各材料间进化树的聚类情况来看,ADH3基因在野生大麦的种系发育上存在较明显的分化。其中,近东野生大麦与西藏野生大麦的亲缘关系更近,而中亚野生大麦与西藏野生大麦的亲缘关系更远。
2.3 群体结构分析
群体结构分析有助于进一步明确不同地理区域野生大麦之间的遗传进化关系,对91份野生大麦的SNP 进行群体结构分析(图3)。设置其群体结构值(K值)为2~4,当K=2 时,91份野生大麦被分为红色和蓝色2个亚群,其中近东和西藏野生大麦以蓝色亚群为主,中亚野生大麦以红色亚群为主;当K=3时,91份野生大麦被分为红色、蓝色和绿色3个亚群,近东野生大麦和西藏野生大麦种质仍以蓝色亚群为主,同时增加了大量绿色亚群的成分,中亚野生大麦仍以红色亚群为主;当K=4 时,91份野生大麦被分为红色、蓝色、绿色和黄色4个亚群,近东野生大麦和西藏野生大麦仍保持相似的群体结构,而中亚野生大麦仍以红色亚群为主。这说明一方面西藏野生大麦的遗传成分与近东野生大麦相近,与系统进化分析得到的结果相一致(图1,2);另一方面也可能是西藏野生大麦和近东野生大麦之间存在互相传播导致基因交流的可能。

图3 野生大麦群体的群体结构(K=2、3、4)Fig.3 Population structure of wild barley populations(K=2,3,4)
3 讨 论
大麦在驯化过程中会经历遗传瓶颈[21-22]和人为干扰[23]等现象,致使其丢失大量的遗传多样性[24],而起源地的野生大麦群体通常表现出较高的遗传多样性[25]。本研究对91份不同来源的野生大麦基于ADH3基因序列的遗传差异分析结果表明,近东野生大麦不仅具有最多的多态性位点个数(S=46)和单倍型个数(H=22),而且近东野生大麦群体的单倍型多样性(Hd=0.914)和核苷酸多样性(π=0.012 65±0.002 47)均明显高于中亚与西藏野生大麦。推测近东野生大麦是栽培大麦的起源中心,而中亚和西藏野生大麦可能是由近东野生大麦扩散后形成的新的多样性驯化中心,所得结论支持近东单起源假说[1,4-9]。
其次,进化和群体结构分析的结果表明3个群体的不同单倍型以近东和西藏野生大麦共有的单倍型Hap9 为中心形成一个大的多样性中心点,且81%的近东野生大麦和80%的西藏野生大麦聚类于Ⅰ类群,91%的中亚大麦与少部分近东和西藏野生大麦混合聚类于Ⅱ类群。群体结构分析也表明西藏野生大麦的遗传成分与近东野生大麦非常相近,表明近东野生大麦和西藏野生大麦群体之间有更近的亲缘关系。推测可能是西藏野生大麦和近东野生大麦之间存在互相传播导致基因交流的可能。这不同于大多数学者认为的近东野生大麦先扩散到中亚后,继续向东由巴基斯坦、印度扩散到中国西藏的路径[1,6],而支持由考古证实的西藏大麦是由近东通过青藏高原以北地区进入东亚,再到达东南部青藏高原的传播途径[17-18]。
本研究通过对不同来源野生大麦材料进行遗传进化分析,有助于进一步了解野生大麦的遗传多样性和群体结构,明确3 个潜在的大麦起源地近东、西藏、中亚野生大麦的遗传进化关系,可为探究大麦的起源提供分子依据,为大麦育种家对种质资源收集、抗性遗传改良等方面的育种工作打下基础。
