食管鳞状细胞癌肿瘤组织内菌群检测及特征分析
2022-06-02冼博鸿位俊敏乔贵宾陈晓辉徐方平骆新兰陈翼翔李子俊
冼博鸿,位俊敏,乔贵宾,陈晓辉,徐方平,骆新兰,陈翼翔,李子俊
(1.南方医科大学第二临床医学院,广东广州 510515;2.广东省人民医院消化内科,广东广州 510080;3.广东省人民医院胸外科,广东广州 510080;4.广东省人民医院病理科,广东广州 510080)
食管鳞状细胞癌是一种发生率高、死亡率高的癌症。2020 年报道食管癌在全球发病率排名第六,死亡率排名第六,5年生存率为20%,仅次于“癌中之王”胰腺癌[1-2]。其中53%的食管癌发生在中国,而在中国90%食管癌是食管鳞状细胞癌[3]。因此,食管鳞状细胞癌是中国特色癌,其中又以山西、广东潮汕、河北、江苏、四川、河南为6 个高发区[4],说明食管鳞癌具有地区特色。目前,食管鳞癌的病因尚未明确,多认为与吸烟、饮酒、生活习惯、饮食习惯相关。近年来,由于生物学高通量测序技术的发展,肿瘤微生态的研究已成为目前热点之一。多项研究表明,不同肠道微生物群的组成能通过影响细胞代谢和免疫功能参与肿瘤的发生发展。类似于幽门螺杆菌引起胃癌,近年来研究发现在多种肿瘤组织及其癌旁正常组织中普遍存在细菌,这些细菌的种类差异与PD-1等免疫治疗疗效相关。而小鼠结直肠癌组织测序研究也表明:原发癌灶和转移灶标本中微生物群保持着一定的稳定性[5]。另外,也有研究发现鼠沙门氏菌[6]、牙龈卟啉单胞菌[7-9]等通过局部炎症反应、调节免疫等途径在胃肠道肿瘤的发展中发挥重要的作用。在食管癌中,最近研究发现食管鳞状细胞癌可能与具核梭杆菌、牙龈卟啉单胞菌感染相关,但缺乏验证,也未对其具体功能及作用途径进行研究[10]。目前食管微生物群落和食管鳞状细胞癌关系的研究样本量均较少,并且只有横断面的研究[11]。因此,有关食管鳞状细胞癌中食管微生物群落分布及功能作用值得深入进一步研究。基于以上发现,我们使用16s rDNA 测序方法研究广东食管鳞癌患者组织中的微生物菌群,期望在食管鳞癌患者癌组织中找出其特征性的细菌,为下一步研究细菌在食管鳞癌发病中的作用提供依据。
1 材料与方法
1.1 研究对象
入选标准:纳入于2014年1月-2015年12月所有就诊于广东省人民医院初诊为食管鳞癌的患者。同期挑选每年体检未发现器质性疾病、口腔疾病、肿瘤家族史的健康人食管组织作为健康对照组。排除标准:①合并其他部位肿瘤、器质性疾病、HBV、HCV 疾病;②手术前4周内有感染性疾病史;③手术前4 周内使用抗生素或PPI 或其他抑酸药物;④手术前4 周内服用激素、调节肠道菌群相关药物,如培菲康、金双歧、适怡等微生态制剂。两组在年龄、性别比例以及民族3 项指标匹配。本课题已经在广东省人民医院(广东省医学科学院)医学研究伦理委员会申请并通过审批,纳入的研究对象均已签署知情同意书。患者和健康受试者随机分为筛查组和验证组。所有食管癌患者组织均经病理活检证实为食管鳞状细胞癌。
1.2 研究方法
1.2.1 组织标本收集 食管癌组织标本来源于广东省人民医院胸外科及病理科,每例标本取癌灶中心组织,用中性缓冲福尔马林液固定,石蜡包埋,连续切片5张,厚度4µm,分别置于涂有防脱片剂的玻片上备用。健康受试者取本院体检对象并行内镜检查者,在内镜下观察食管黏膜正常的中段位置,活检组织两块,用中性缓冲福尔马林液固定后立即存放于-80 ℃冰箱保存。
1.2.2 DNA 提取及储存 使用UltraClean®MicrobialDNA Isolation Kit(美国,MOBI 公司)试剂从组织切片中提取细菌DNA,具体提取方法按照试剂盒说明书进行。收集滤液,保存在-20 ℃冰箱。要求DNA 总量≥150 ng;DNA 浓度≥5 ng/µL;有明显主带,无降解、无RNA和蛋白质等杂质污染。
1.2.3 16s rDNA 测序和数据处理 对合格的DNA样品取适量置于离心管中,根据V4 区使用带Barcode的引物515F和806R,进行PCR,PCR 在缓冲液中进行,另外加入2×Premix Taq 引物酶及dNTP,然后进行热循环,在94 ℃条件下变性5 min,再进行30 个扩增循环(包括94 ℃变性30 s,52 ℃退火30 s及72 ℃延伸30 s),再于72 ℃充分延伸10 min。最后置于4 ℃保存。用1%琼脂糖凝胶电泳检测PC产物的片段长度和浓度,主带长度在正常范围内的样品可用于进一步的研究。利用GeneT-ools Analysis Software 对PCR 产物进行浓度对比后,按照等质量原则计算各样品所需体积,将各PCR产物进行混合。使用E.Z.N.A.GelExtractionKit 凝胶回收试剂盒回收PCR 混合产物,TE 缓冲液洗脱回收目标DNA片段。按照NEBNextUltraD-NA Library Prep Kit for Illumina®标准流程进行建库操作。使用Illumina Hiseq2500 平台对构建的扩增子文库进行PE250 测序。利用Trimmomatic 软件对测序数据进行Paired-end raw Reads 过滤,利用FLASH 对每对PE reads 进行拼接,利用Mothur 软件进行Raw Tags 序列质量过滤,最终得到有效的拼接片段(Clean Tags)。
1.2.4 生物信息学分析 使用UPARSE软件(V10)将所有样品的全部cleantag 进行聚类,以97%的一致性将序列聚类成OTUs(operational taxonomic units)。将其中具有代表性的序列进行物种分类注释,最后进行Alpha 多样性分析、Beta 多样性分析及LEfSe分析。
1.2.5 Q-PCR 方法验证靶细菌 通过Q-PCR 检测靶细菌菌的表达水平。根据筛选出来的靶细菌设计特异性引物序列,用紫外分光光度计测定阳性重组质粒的浓度。然后将质粒稀释10 倍,分别用10-2、10-3、10-4、10-5、10-6、10-7等浓度的样品绘制标准曲线,然后作为DNA 标准品置于-20 ℃保存。将SYBR Mix10 µL、上游引物0.6 µL、下游引物0.6µL、DNA模板1 µL混合,加入无菌水至共20µL。上机后第一步在95 ℃下反应30 s 共1 个循环,第二步在95 ℃下反应5 s 后在60 ℃下反应30 s共40 个循环。最后将每份样本的DNA 稀释到100 ng/µL 下检测DNA 的含量,每次样品检测重复3 次,取平均值,通过标准曲线计算目标细菌在样品中的表达量,以拷贝数表示。
1.3 统计学方法
采用SPSS 22.0 软件进行统计分析,P<0.05 认为差异有统计学意义。采用Mann-WhitneyU检验比较细菌相对丰度的差异,差异采用独立样本t检验比较年龄和细菌拷贝数量的差异,采用卡方检验检测性别、吸烟、饮酒、拷贝数的差异,使用对数转换使拷贝数具有方差齐性。LEfSe分析上首先采用多组比较的秩和检验检测不同分组间丰度差异显著的物种,有统计学意义的组间进行两两比较,最后用线性判别分析(LDA)来实现降维和评估差异显著物种的影响大小(LDA Score),设置该大小的筛选值≥2。利用SPSS 22.0软件绘制ROC曲线。
2 结果
2.1 患者情况
本研究纳入ESCC 患者共105 例,健康对照组共54 例,均为汉族,其中筛选组纳入ESCC 患者42例,健康对照组37例;验证组纳入ESCC患者63例,健康对照组17例。
2.2 16SrDNA测序筛选靶细菌
2.2.1 OTU 构建及物种分析 通过对筛选组样品提取的序列进行拼接,过滤掉低质量序列后,ESCC组和健康对照组的组织样本共获得29 281 618 条序列,平均每个样本为532 393条。ESCC 组获得序列平均为523 243 条,健康对照组获得序列平均为543 372条。以97%的一致性将序列聚类为OTU。
对样本OTU 聚类进行分析,在phylum(门)和genus(属)水平统计分析各样本的群落组成。物种分析表明,在门水平上,两组相对丰度≥1%的优势菌门相同,均为放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)变形菌门(Proteobacteria)、螺旋体门(Spirochaetes),虽然两组的物种组成差异不大,但丰度比例却不相同。ESCC组与HC组相比,厚壁菌门、拟杆菌门及梭杆菌门丰度升高,而放线菌门及变形菌门丰度明显降低(图1)。
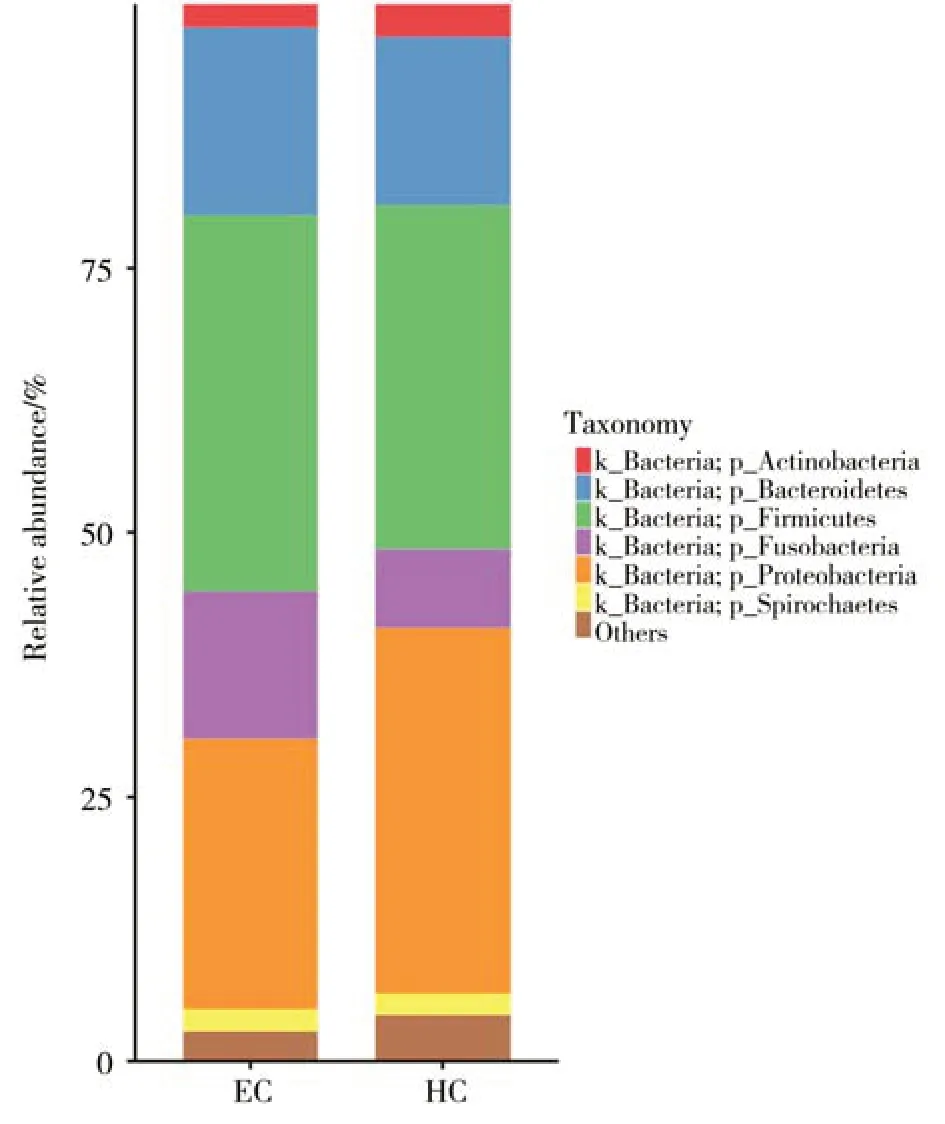
图1 ESCC组和HC组在门水平的相对丰度柱形图Fig.1 Histogram of relative abundance of ESCC group and HC group at phylum level
在属水平,ESCC 组以梭杆菌属(Fusobacterium)、孪生球菌属(Gemella)、奈瑟菌属(Neisseria)、卟啉单胞菌属(Porphyromonas)、链球菌属(Streptococcus)为主,健康对照组以梭杆菌属(Fusobacterium)、放线杆菌属(Actinobacillus)、链球菌属(Streptococcus)、普雷沃氏菌属(Prevotella)为主,其中相比于健康对照组,ESCC 组放线杆菌属(Actinobacillus)、梭杆菌属(Fusobacterium)、微单胞菌属(Parvimonas)、嗜血杆菌属(Haemophilus)、奈瑟菌属(Neisseria)、卟啉单胞菌属(Porphyromonas)、链球菌属(Streptococcus)、韦荣氏球菌属(Veillonella)丰度明显升高,嗜盐单胞菌属(Halomonas)、罗伊氏乳杆菌属(Lactobacillus)、棒状杆菌属(Oribacterium)、月单胞菌属(Selenomonas)、普雷沃氏菌属(Prevotella)丰度明显降低(图2)。

图2 ESCC组和HC组在属水平的相对丰度柱形图Fig.2 Histogram of relative abundance of ESCC group and HC group at genus level
2.2.2 多样性分析 通过α 多样性及β 多样性评估两组样本序列中物种多样性差异。α多样性(Alpha diversity)是对单个样品中物种多样性的分析,统计参数包括Observedspecies 指数、Chao1 指数、Shannon 指数、Simpson 指数以及PD wholetree 等。Chao1和Observedspecies 数值表示所测样品中群落的丰度,其值越高,则物种丰度越高,经统计学分析两组物种丰度差异无统计学意义(P>0.05)。Shannon 指数、simpson 指数以及PD_whole_tree 值代表群落多样性程度,其值越高,则多样性越高,差异均无统计学意义(表1)。
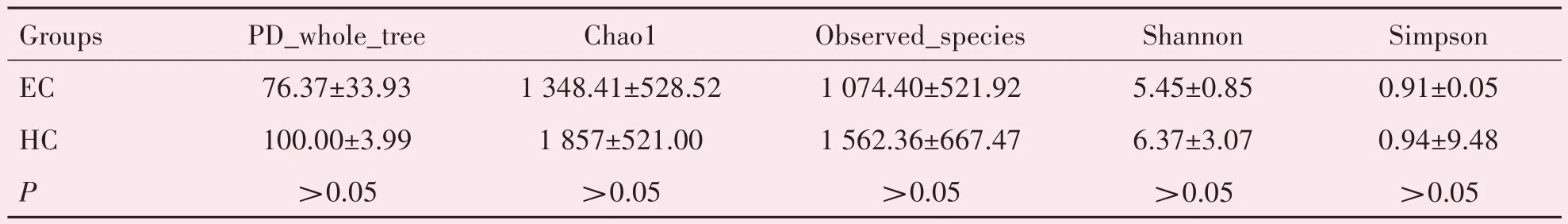
表1 ESCC组和HC组样本α多样性参数比较Table 1 Comparison of alpha diversity parameters between ESCC group and HC group
通过分析样本序列的β 多样性(Beta Diversity)比较两组样本间微生物群落构成的差异,统计所有OTU 在不同样品中的相对丰度,之后利用Bray-Curtis 距离公式计算不同样品间的间相异系数矩阵,对矩阵进行层级聚类及样本距离heatmap图,颜色越红表明样本间距离越远。在绘制出的heatmap图中,我们可以发现ESCC 组与HC 组对比样本距离远,ESCC 组中各样本的平均样本距离为0.502,而EC 组与HC 组中各样本的平均距离为0.685,说明两组细菌群落构成存在差异(图3)。
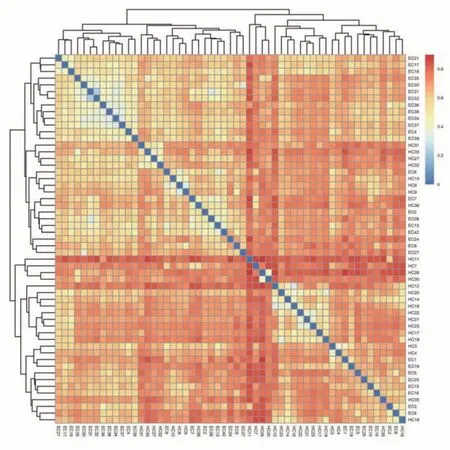
图3 ESCC组和HC组样本距离heatmap图Fig.3 Heatmap of sample distance between ESCC group and HC group
2.2.3 LEfSe 分析 最后,使用LEfSe 分析寻找两组之间具有统计学差异的物种。LEfSe 分析得出ESCC 组中明显升高的差异菌群共有22 种,其中包括卟啉单胞菌属、链球菌属、梭杆菌属在内的12 种菌在ESCC组中显著增多(P<0.05),而在HC组中,包括极毛杆菌目、毛螺菌科、肠杆菌目等10 种菌明显升高(P<0.05;图4)。

图4 部分序列LDA值分布柱状图Fig.4 Histogram of partial sequence LDA value distribution
2.3 Q-PCR方法验证靶细菌
根据筛选结果,发现卟啉单胞菌属、梭杆菌属在ESCC 组样本中显著增高,后选择该菌属中既往研究有意义的具核梭杆菌和牙龈卟啉单胞菌作为靶细菌,用Q-PCR 测序的方法验证筛选结果。对不符合正态分布的拷贝数数据进行对数转换,其中ESCC 患者和健康人具核梭杆菌和牙龈卟啉单胞菌的平均拷贝数及对数转换后数据分别见表2-3。并以此绘制ROC曲线,验证ESCC患者和健康人群中具核梭杆菌和牙龈卟啉单胞菌的拷贝数差异,结果显示,与健康对照组相比,ESCC 组牙龈卟啉单胞菌及具核梭杆菌的拷贝数显著升高(P<0.05)。绘制的ROC曲线中,牙龈卟啉单胞菌、具核梭杆菌的AUCs分别为0.902、0.708,与截断值相关的敏感性和特异性分别为80.0%、5.9%以及45.8%,10.0%(图5)。
表2 ESCC组与EC组牙龈卟啉单胞菌及具核梭杆菌拷贝数Table 2 Copy numbers of Porphyromonas gingivalis and Fusobacterium nucleatum in ESCC group and EC group()
EC:esophageal cancer;HC:healthy control.
3 讨论
食管癌特别是食管鳞癌在中国发病率最高,也成为我国肿瘤疾病中影响国人健康的重要负担之一。目前食管癌的病因尚未明确,针对病因的治疗和预防措施乏力[12]。近来研究表明,组织微环境中的胞内菌在癌症发病及治疗应答中起重要作用[13]。Binder Gallimidi等[14]报道在4-硝基喹啉-1-氧化物(4NQO)诱导口腔鳞癌模型中发现牙龈卟啉单胞菌感染能够刺激口腔上皮细胞的TLR 受体,激活IL-6/STAT3 信号通路,进而促进口腔鳞状细胞癌生长。另外,有学者发现具核梭杆菌能通过调节结直肠癌组织内T 细胞的分布、PD-L1 的表达影响肿瘤的发生发展[15-16]。而使用抗生素杀灭具核梭杆菌后,则可以抑制结肠癌细胞的生长[17]。王晴萱等[18]经牙龈卟啉单胞菌和具核梭杆菌单独或联合感染人口腔表皮样癌细胞(KB 细胞),发现牙龈卟啉单胞菌感染KB细胞能抑制细胞周期,促进KB细胞分泌IL-6 和IL-8,引起促进口腔鳞状细胞癌生长和引起免疫炎症反应。这些研究均表明牙龈卟啉单胞菌、具核梭杆菌在肿瘤发病中的作用。
表3 ESCC组与EC组牙龈卟啉单胞菌及具核梭杆菌对数转换后Table 3 Copy numbers of Porphyromonas gingivalis and Fusobacterium nucleatum in ESCC group and EC group()

表3 ESCC组与EC组牙龈卟啉单胞菌及具核梭杆菌对数转换后Table 3 Copy numbers of Porphyromonas gingivalis and Fusobacterium nucleatum in ESCC group and EC group()
EC:esophageal cancer;HC:Healthy control.
目前对于食管癌组织菌群的研究报道较少,我们在前期研究中通过对食管癌患者口腔唾液研究发现食管鳞癌患者唾液中牙龈卟啉单胞菌属相对于健康对照组表达明显升高[19]。并且,在体外细胞实验中,我们发现牙龈卟啉单胞菌能通过激活NFkB信号通路促进食管鳞癌进展[20]。
基于以上研究,我们使用16s rDNA 测序方法检测了食管鳞癌患者组织中的菌群谱,通过α 多样性参数分析比较发现ESCC组与健康对照组在各参数差异没有统计学意义,提示两组菌群多样性上没有显著差异。但在物种分析上,我们发现在ESCC组中以梭杆菌属(Fusobacterium)、孪生球菌属(Gemella)、奈瑟菌属(Neisseria)、卟啉单胞菌属(Porphyromonas)、链球菌属(Streptococcus)为主,LEfSe分析表明ESCC组中卟啉单胞菌属、链球菌属、梭杆菌属丰度显著升高,且差异且有统计学意义。
此外,我们还首次通过Q-PCR 方法分析发现食管癌患者组织内菌群构成与健康对照组有明显差异,菌群组成具有特征性。ESCC 组卟啉单胞菌属、梭杆菌属相对于同组其他菌属来讲表达明显升高,以牙龈卟啉单胞菌、具核梭杆菌为代表,且差异明显。前期我们在食管癌口腔唾液研究中也发现具核梭杆菌和牙龈卟啉单胞菌是口腔的常见菌。以往有研究表明牙龈卟啉单胞菌在食管鳞状细胞癌组织中增多,本研究进一步细化并发现具核梭杆菌、牙龈卟啉单胞菌均为食管鳞状细胞癌的特征性菌群,在致癌机制上可能存在相互促进的作用。
具核梭杆菌和牙龈卟啉单胞菌二者均为革兰氏阴性严格厌氧杆菌,可释放多种毒力因子,是重要牙周病致病菌。有研究还发现具核梭杆菌可以提高牙龈卟啉单胞菌对上皮细胞的黏附和入侵,进而加强牙龈卟啉单胞菌的致病性[21-22]。我们在前期研究中,也发现具核酸杆菌联合牙龈卟啉单胞菌共感染食管癌细胞可上调PD-L1表达水平。此外,也有其他研究报道牙龈卟啉单胞菌细胞壁成分肽聚糖被细菌外膜囊泡吸收入细胞,内化肽聚糖触发胞质受体通过RIP2 依赖的方式诱导PD-L1 在口腔癌中的表达[23]。这些研究依据表明牙龈卟啉单胞菌、具核梭杆菌在食管鳞状细胞癌中富集,可能对食管鳞状细胞癌具有促进其恶性行为及对免疫治疗疗效可能起调节作用。因此,下一步我们将对牙龈卟啉单胞菌和具核梭杆菌联合调节食管鳞状细胞癌PD1免疫治疗应答的机制作进一步深入研究。研究其致癌及诱导免疫耐药的机制,结果为食管鳞状细胞癌筛选的生物靶标及提高临床免疫治疗效果作有益探索。
