Prognostic potential of the small GTPase Ran and its methylation in hepatocellular carcinoma
2022-06-02HuiHuiLiuJuWngYingZhngYuChenFnKiWng
Hui-Hui Liu , Ju Wng , Ying Zhng , Yu-Chen Fn , b , c , Ki Wng , b , c , *
a Department of Hepatology, Qilu Hospital of Shandong University, Jinan 250 0 0 0, China
b Shenzhen Research Institute of Shandong University, Shenzhen 5180 0 0, China
c Institute of Hepatology, Shandong University, Jinan 250 0 0 0, China
Keywords:Hepatocellular carcinoma Ran Methylation Prognosis Immune cell infiltration
ABSTRACT
Introduction
Hepatocellular carcinoma (HCC) comprises 75%-85% of primary liver cancer, and shares several defining characteristics, such as rising global incidence, limited treatment options and high mortality [1] . Most HCC patients are diagnosed at advanced stages due to asymptomatic presentations of early stages and the lack of effective biomarkers [ 2 , 3 ]. With a 5-year relative survival rate of 18%,liver cancer has been ranked second after pancreatic cancer [4] .The identification of robust biomarkers, which can be employed for predicting prognosis of HCC, is therefore mandatory.
DNA methylation serves as a covalent modification and occurs on cytosine nucleotides in the context of CpG dinucleotides [5] .Researches have demonstrated that DNA methylation is closely related to the occurrence, diagnosis, prognosis and treatment of liver cancer. Aberrant DNA methylation patterns play important roles in the early occurrence and development of HCC [6] . The average methylation level of alpha-1A adrenergic receptor (ADRA1A) gene could distinguish HCC tissues from nontumor tissues, suggesting that it is an effective biomarker for the diagnosis of HCC [7] .The combination of epigenetic therapy and immunotherapy significantly enhances tumor regression compared with epigenetic therapy or immunotherapy alone [8] . Multiple studies have individually recognized Ras superfamily members as critical regulators in cancers. The Rho protein is a member of the Ras superfamily, and DNA methylation of Rho GTPase activating protein 30 (ARHGAP30)is negatively correlated with ARHGAP30 gene expression in lung adenocarcinoma [9] . Rab39a, another member of Ras superfamily,is identified as a potential epigenetic silenced tumor suppressor inhibiting cervical cancer invasion and migration [10] . Particularly,Ras proximate protein 2A (RAP2A) expression is correlated with its methylation and emerged as an independent marker for prognosis of HCC patients [11] . Ras-related nuclear protein (Ran), which belongs to the Ras superfamily, plays a distinct role in cellular processes involving the regulation of nucleocytoplasmic transport of macromolecules and the control of cell cycle progression [ 12 , 13 ].Emerging evidence has revealed that the upregulation of Ran plays an oncogenic role in tumors and correlates with poor prognosis.For instance, Ran was significantly overexpressed in human colorectal carcinoma (CRC) tissues and cell lines, which drives the development and progression of CRC [14] . Ran served as a cancer driver gene (CDG) for malignant pleural mesothelioma (MPM), and the elevated expression of Ran impacts pleural tumorigenesis [15] .A recent study demonstrated that upregulated Ran was positively correlated with a low overall survival (OS) rate in patients with breast cancer [16] . Of particular note, Ran (rs14035) was associated with the survival of HCC patients [17] . Although the Ran expression patterns have been dissected in several tumors, the clinical significance and prognostic value of Ran expression and its promoter methylation in HCC are poorly understood.
The present study aimed to investigate the prognostic roles of Ran mRNA expression and its methylation in HCC patients.
Methods
Data sources
The Cancer Genome Atlas (TCGA) was used to explore the mRNA expression of Ran between HCC and normal tissues. The samples with complete clinical and transcriptional data were included. Gene names were mapped to an annotation file of Ensembl(http://www.ensembl.org/index.html), and the Ran expression levels were identified by employing “limma” R package. The potential relationship between Ran mRNA expression and its promoter methylation was extensively exploited, and their prognostic roles were also evaluated in HCC.
Correlation analyses of Ran with immune cells and genes
Five types of tumor-infiltrating immune cells, including B cells,CD4 + T cells, myeloid dendritic cells, macrophages and neutrophils, were downloaded from Tumor Immune Estimation Resource (TIMER) (http://timer.cistrome.org/) which allowed us to visualize relationships of Ran expression with immune infiltration levels in HCC.
Immune-related genes were extracted from the IMMUNE_SYSTEM_PROCESS gene set (systematic name: M13664)and IMMUNE_RESPONSE gene set (systematic name: M19817)in Gene Set Enrichment Analysis (GSEA) (https://www.gseamsigdb.org/gsea/index.jsp). We identified the expression levels of immune-related genes using “limma” R package, and calculated average values of genes that matched multiple probes.Then univariate Cox analysis was applied to identify prognostic immune-related genes. Based on the TIMER website, we further evaluated Ran mRNA expression with prognostic immune-related genes in HCC by employing Spearman rank correlation analysis.
Functional analysis of differentially expressed genes (DEGs) in the high and low Ran expression groups
According to the median value of the Ran mRNA expression levels, HCC patients were subdivided into the high Ran expressiongroup and the low Ran expression group. Based on the screening criteria of a |log 2 fold change| (|log 2 FC|)>1 and a false discovery rate (FDR)<0.05, we screened out differentially expressed genes by employing “limma” package in R software. Applying “clusterProfiter” R package, we conducted gene functional enrichment analysis, comprising gene oncology (GO) functional annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway analysis.
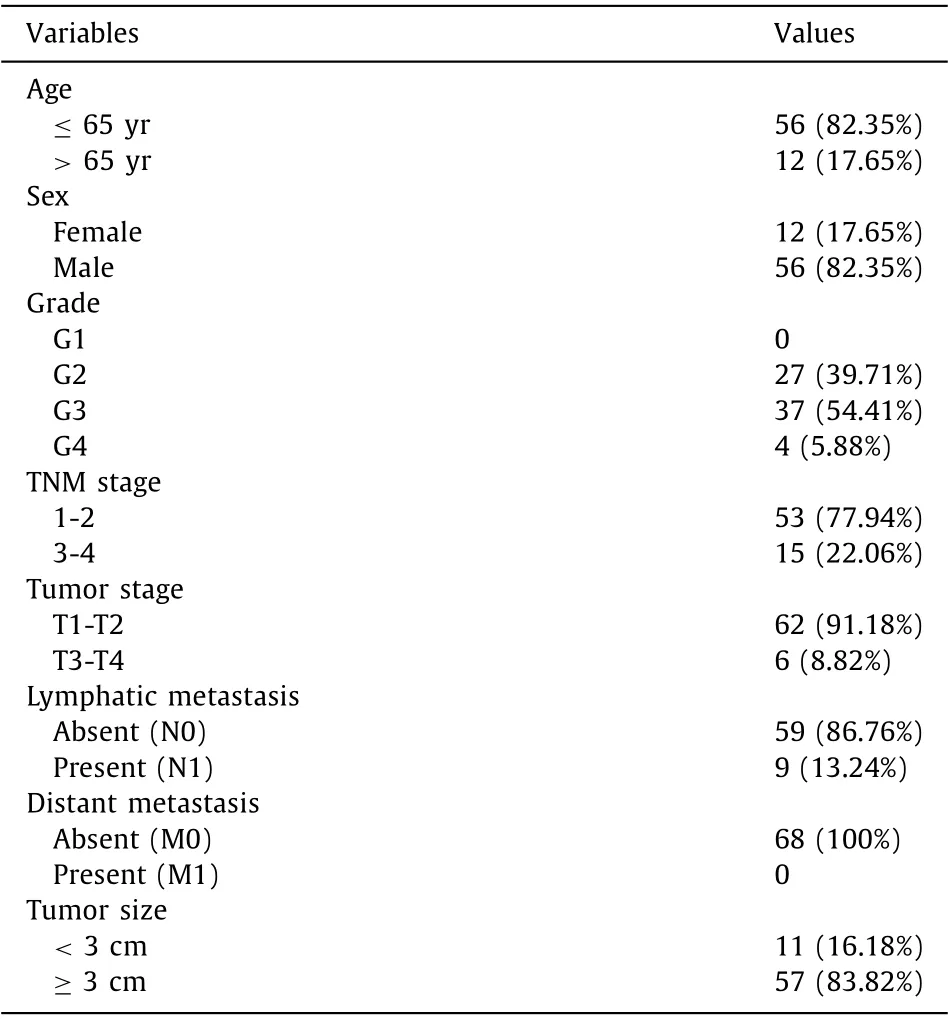
Table 1 Baseline characteristics of enrolled patients with hepatocellular carcinoma( n = 68).
Experimental verification of Ran differential expression in HCC tissues
We obtained 68 primary HCC tissues and 64 normal tissues at our hospital between December 2019 and November 2020. We validated Ran mRNA expression levels in HCC tissues and paired adjacent nontumor tissues. The study was approved by the Ethics Committee of Qilu Hospital of Shandong University (No. 2018 Scientific research express 515). All participants or their relatives signed written informed consent before enrolling in the study. All HCC patients underwent curative surgical resection and were pathologically confirmed. The following patients were excluded: patients with history of other tumors, patients previously received transarterial chemoembolization (TACE), microwave ablation (MWA), and chemotherapy, and those who suffered from metastatic diseases.All tissues were snap-frozen in liquid nitrogen until total RNA isolation. Baseline characteristic of HCC patients enrolled in the study are summarized in Table 1 .
Total RNA was extracted from samples by using TRIzol reagent(Invitrogen, Carlsbad, CA, USA) and resuspended in 20 μL RNasefree water. RNA was converted into complementary DNA then immediately used for analysis or stored at -70 °C. Using quantitative real time polymerase chain reaction (qRT-PCR), the Ran mRNA expression levels were detected with SYBR Premix Ex Taq (Takara,Shiga, Japan).β-actin was used as an internal control. The PCR primers were as follows: Ran, 5 ′ -GGT GGT ACT GGA AAA ACG ACC-3 ′ (forwards) and 5 ′ - CCC AAG GTG GCT ACA TAC TTC T-3 ′ (reverse); andβ-actin, 5 ′ -AAG GTG ACA GCA GTC GGT T-3 ′ (forwards)and 5 ′ -TGT GTG GAC TTG GGA GAG G-3 ′ (reverse) [ 18 , 19 ]. The 2-ΔΔCtmethod was used to determine the relative expression of Ran andβ-actinmRNA.
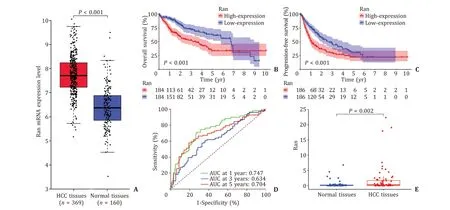
Fig. 1. Ran expression in HCC tissues and normal tissues. A : Ran mRNA was overexpressed in HCC tissues in TCGA database; B and C : Kaplan-Meier curves for HCC patients stratified according to the median Ran mRNA expression levels; D: time-dependent ROC curves for Ran expression at 1-, 3-, and 5-year in HCC patients; E: Ran mRNA was overexpressed in HCC tissues in experimental analysis. AUC: area under the receiver operating characteristic curve.
Statistical analysis
All results were produced with R software (Version 4.0.2) in our present study. Kaplan-Meier curves and log-rank tests were used to estimate the prognostic potential of Ran expression along with its promoter methylation. Time-dependent receiver operating characteristic (ROC) curves were applied to evaluate the predictive performance of Ran in predicting overall survival (OS). Univariate and multivariate Cox analyses were employed to explore whether Ran expression was an independent prognostic factor in HCC patients.We adopted Spearman rank correlation analysis to assess the relationships between Ran expression and its promoter methylation.Chi-square test was used to appraise clinical significance of Ran expression and its promoter methylation. AllPvalues were two tailed and less than 0.05 were considered statistically significant.
Results
Ran expression in HCC tissues and normal tissues
A total of 374 HCC tissues and 50 normal tissues with Ran expression were extracted from TCGA, and OS (n= 368 HCC patients) and progression-free survival (PFS) (n= 372 HCC patients)were collected. We found that Ran mRNA expression level was significantly increased in HCC tissues compared with normal tissues (P<0.001, Fig. 1 A). Kaplan-Meier curves and log-rank tests showed that the OS rate of the high Ran expression group was significantly lower than that of the low Ran expression group(P<0.001, Fig. 1 B), and the PFS of the low Ran expression group was significantly higher than that of the high Ran expression group(P<0.001, Fig. 1 C).
Time-dependent ROC curves were plotted to evaluate the predictive ability of Ran expression, and the results suggested that the area under the receiver operating characteristic curve (AUC) values at 1-, 3-, and 5-year were 0.747, 0.634 and 0.704, respectively( Fig. 1 D), validating the potential significance of Ran expression in the prognosis of HCC patients. Univariate Cox analysis showed that T stage 3-4, M1 stage, TNM stage 3-4 and Ran expression were correlated to poor prognosis of HCC (allP<0.05, Table 2 ), whereas multivariate Cox analysis identified that only Ran expression was an independent prognostic factor for HCC patients (HR = 1.492,95% CI: 1.129-1.971,P= 0.005; Table 2 ).
Consistent with TCGA data, our qRT-PCR found that Ran expression was significantly higher in HCC tissues than in normal tissues(P= 0.002, Fig. 1 E).
Association between Ran expression and its promoter methylation
A total of 430 HCC patients with Ran methylation were extracted from TCGA. The ranked distribution of Ran promoter CpG sites is shown in Fig. 2 A. Taking the average methylation level of all CpG sites, we obtained the methylation level of Ran, which was negatively correlated with its expression (r= -0.36,P<0.001, Fig. 2 B). A detailed description of the associations between methylation levels of CpG sites and Ran expression is provided in Fig. 2 C-G. Ran expression was negatively related with methylation levels of cg01060409 (r= -0.26,P<0.001), cg17629322 (r= -0.36,P<0.001), cg25201101 (r= -0.33,P<0.001), cg01758167 (r= -0.16,P<0.05) and cg19847963(r= -0.23,P<0.001).
According to the median methylation levels of these CpG sites,the patients were divided into the hypermethylated group and hypomethylated group. A total of 424 HCC patients with OS and 428 HCC patients with PFS were obtained from TCGA. We observed that the hypermethylated CpG sites cg17629322 and cg25201101 were associated with favorable OS (P= 0.0 03 andP= 0.0 06, Fig. 3 A, B),and the hypermethylated cg01060409 and cg19847963 were correlated with favorable PFS (P= 0.040 andP= 0.020, Fig. 3 C, D).
Clinical significance of Ran expression and its methylation
As shown in Table 3 , Ran expression was significantly different in terms of grade (P<0.001), TNM stage (P= 0.037), T stage (P= 0.025) and Ran methylation (P<0.001), whereas the methylation level of Ran was affected only by Ran mRNA expression (P<0.001). Adopting “stat_compare_means” in R software, we performed subgroup analyses based on Ran mRNA expression and promoter methylation. The grade 1 patients demonstrated lower Ran mRNA expression than the grade 3 and grade 4 patients (P<0.001,P<0.05, respectively). Similarly, the Ran mRNA expression levels of the grade 3 and 4 patients were significantly increased compared with those of the grade 2 patients(P<0.001,P<0.05, respectively; Fig. 4 A). Fig. 4 B reveals that Ran mRNA expression levels in patients with stage I were lower than those in patients with stage II and stage III (P= 0.014,P<0.001,respectively). An analogous trend was observed when comparing Ran mRNA expression in patients with T1 stage, T2 stage and T3 stage (P<0.05,P<0.001, Fig. 4 C).
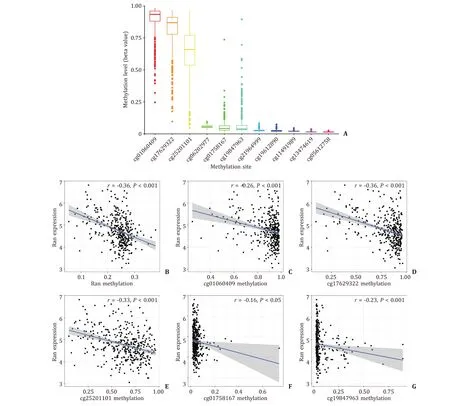
Fig. 2. Ran promoter methylation in HCC patients. A : The distribution of 11 Ran promoter CpG sites; B : Ran expression was negatively correlated with its promoter methylation; C - G : negative correlation between Ran expression and CpG sites. HCC: hepatocellular carcinoma.
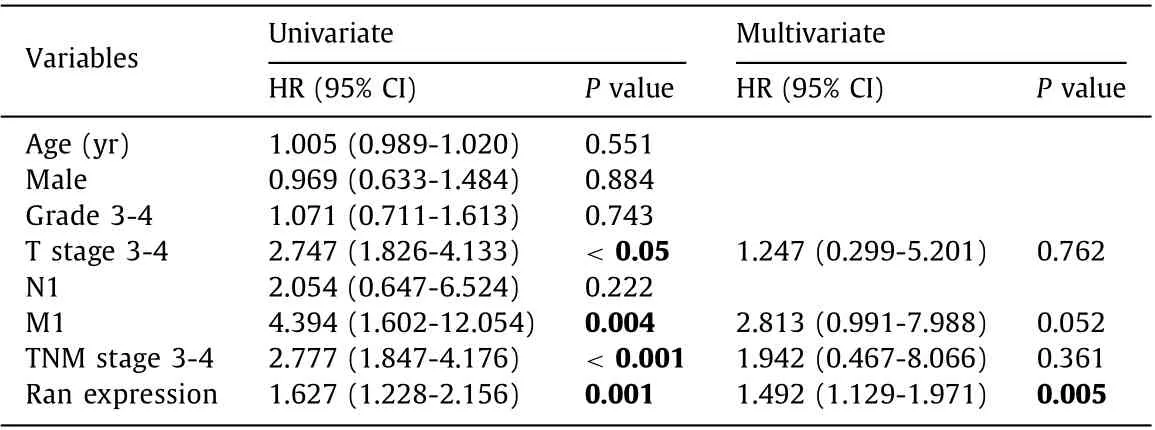
Table 2 Uni- and multivariate Cox regression analyses of prognosis factors associated with survival in patients with hepatocellular carcinoma.
The methylated Ran promoter levels were higher in HCC patients aged>65 years (P= 0.041, Fig. 4 D). Compared with the stage II and stage IV patients, the stage I patients showed hypermethylation of Ran promoter (P= 0.030 andP= 0.026, Fig. 4 E).We also found that the methylation levels of Ran promoter in patients with T1 stage was higher compared with those in patients with T2 and T4 stages (P= 0.049 andP= 0.048,Fig. 4 F).
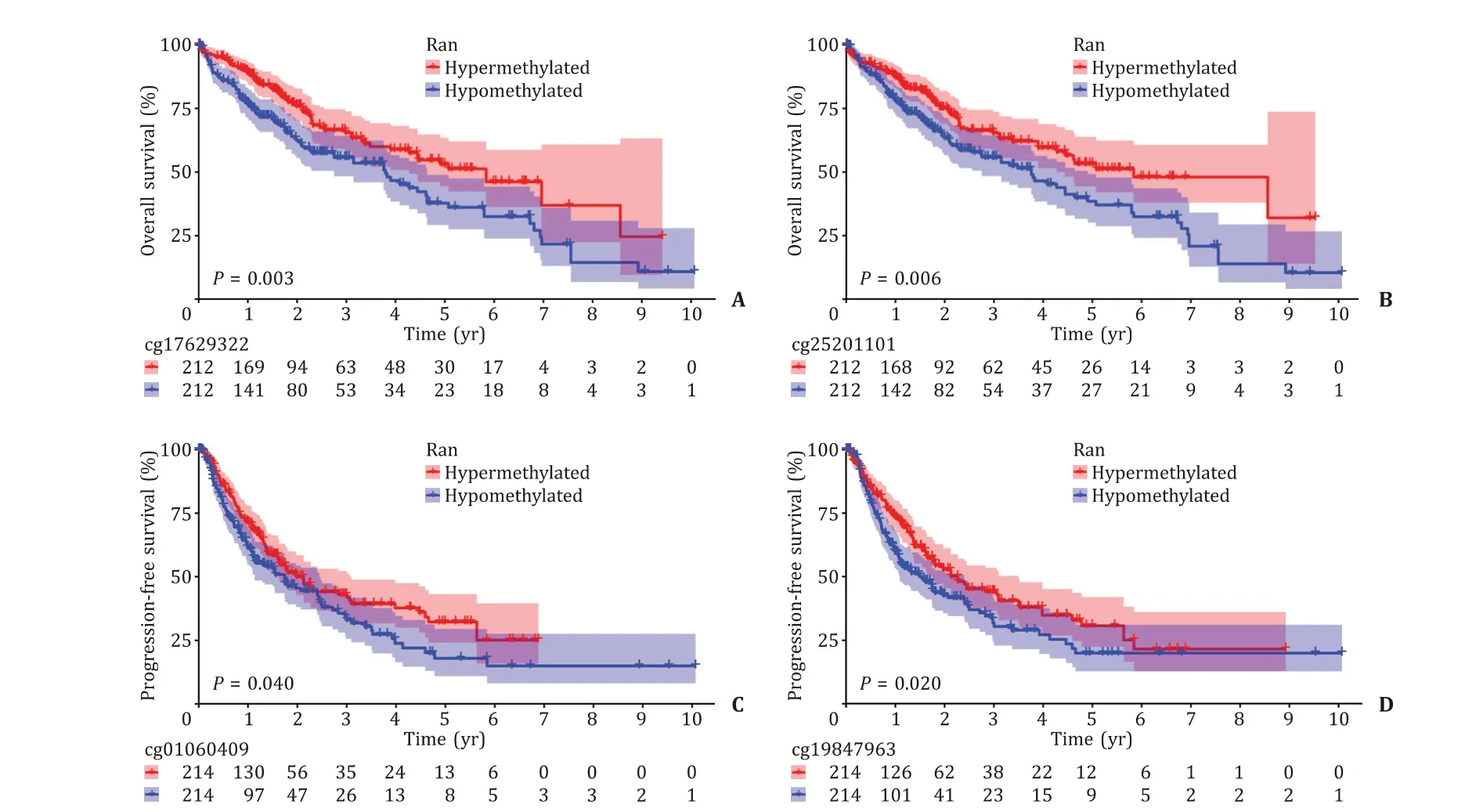
Fig. 3. Kaplan-Meier curves of hypermethylated and hypomethylated Ran promoter CpG sites. A: cg17629322; B: cg25201101; C: cg01060409; D: cg19847963.
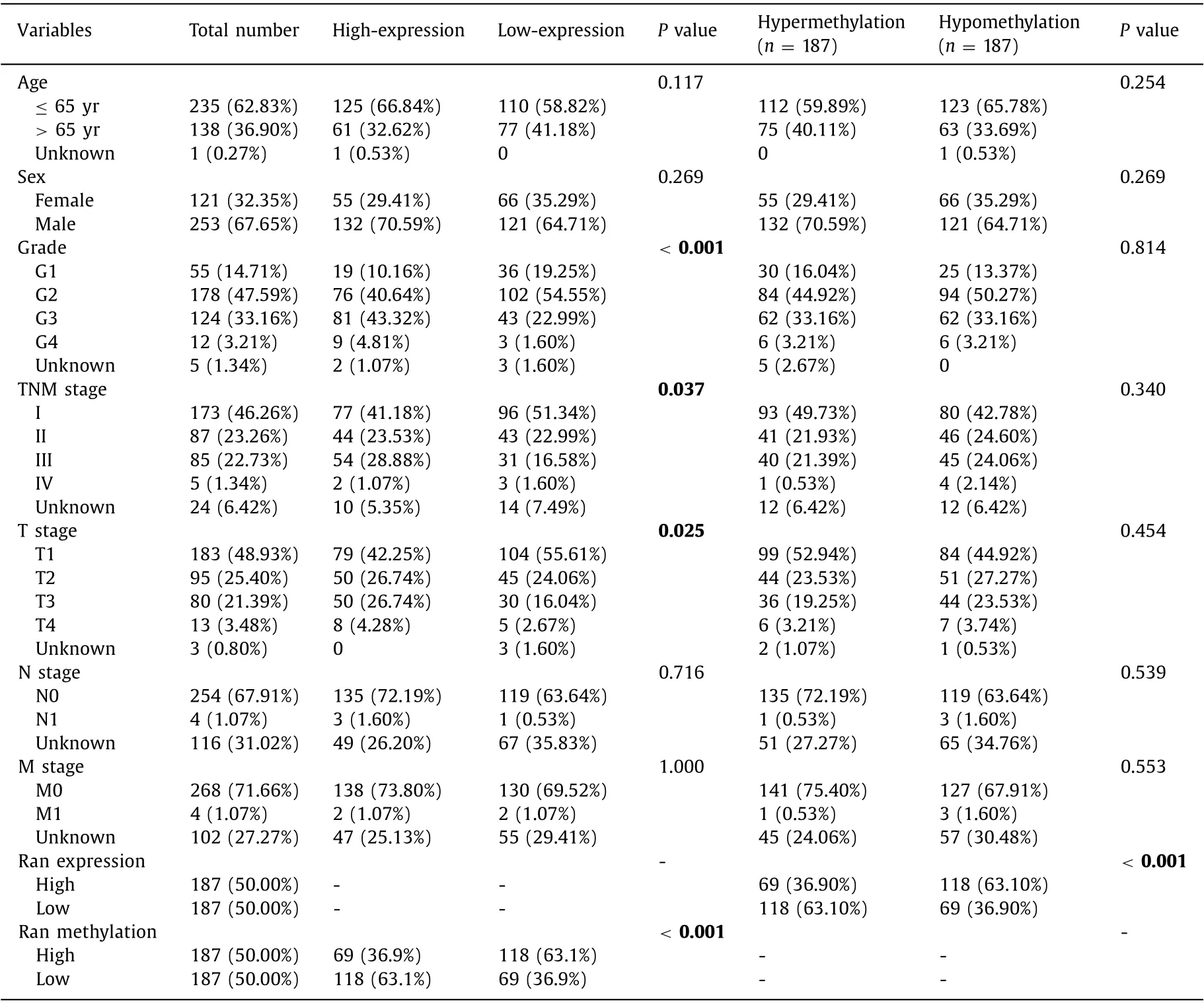
Table 3 Correlation between Ran mRNA expression/methylation and clinicopathological features in TCGA database.
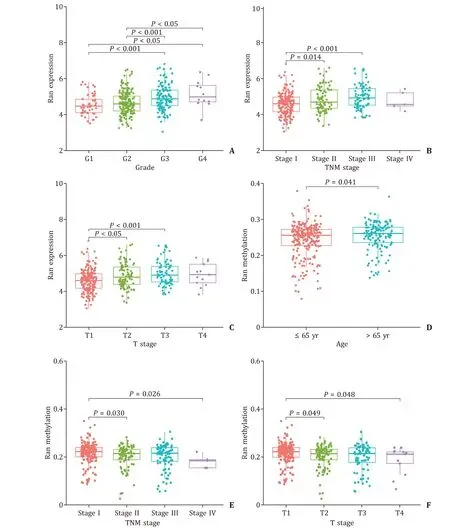
Fig. 4. The correlations between Ran expression/promoter methylation and clinical characteristics in TCGA database. A - C : The relationships between Ran expression and grade, TNM stage and T stage in HCC patients; D - F : the relationships between Ran promoter methylation and age, TNM stage and T stage in HCC patients. G: grade; T: tumor stage; HCC: hepatocellular carcinoma.
Association between Ran mRNA expression and immune infiltrates or immune-related genes
As shown in Fig. 5 A-E, the correlations between Ran mRNA expression and B cells, CD4 + T cells, myeloid dendritic cells,macrophages and neutrophils were 0.409, 0.260, 0.531, 0.408 and 0.230, respectively (AllP<0.001).
In total, 330 immune-related genes were obtained from GSEA database, and univariate Cox analysis showed that 36 of them were prognostic immune-related genes. Based on the TIMER website, we finally identified the expression levels of 32 prognostic immune-related genes. Therefore, we analyzed the correlation between Ran expression and the expression of 32 immunerelated genes. Aside fromAQP9andIL27, other prognostic immunerelated genes presented positive correlations with Ran expression(allP<0.001, Fig. 6 ).
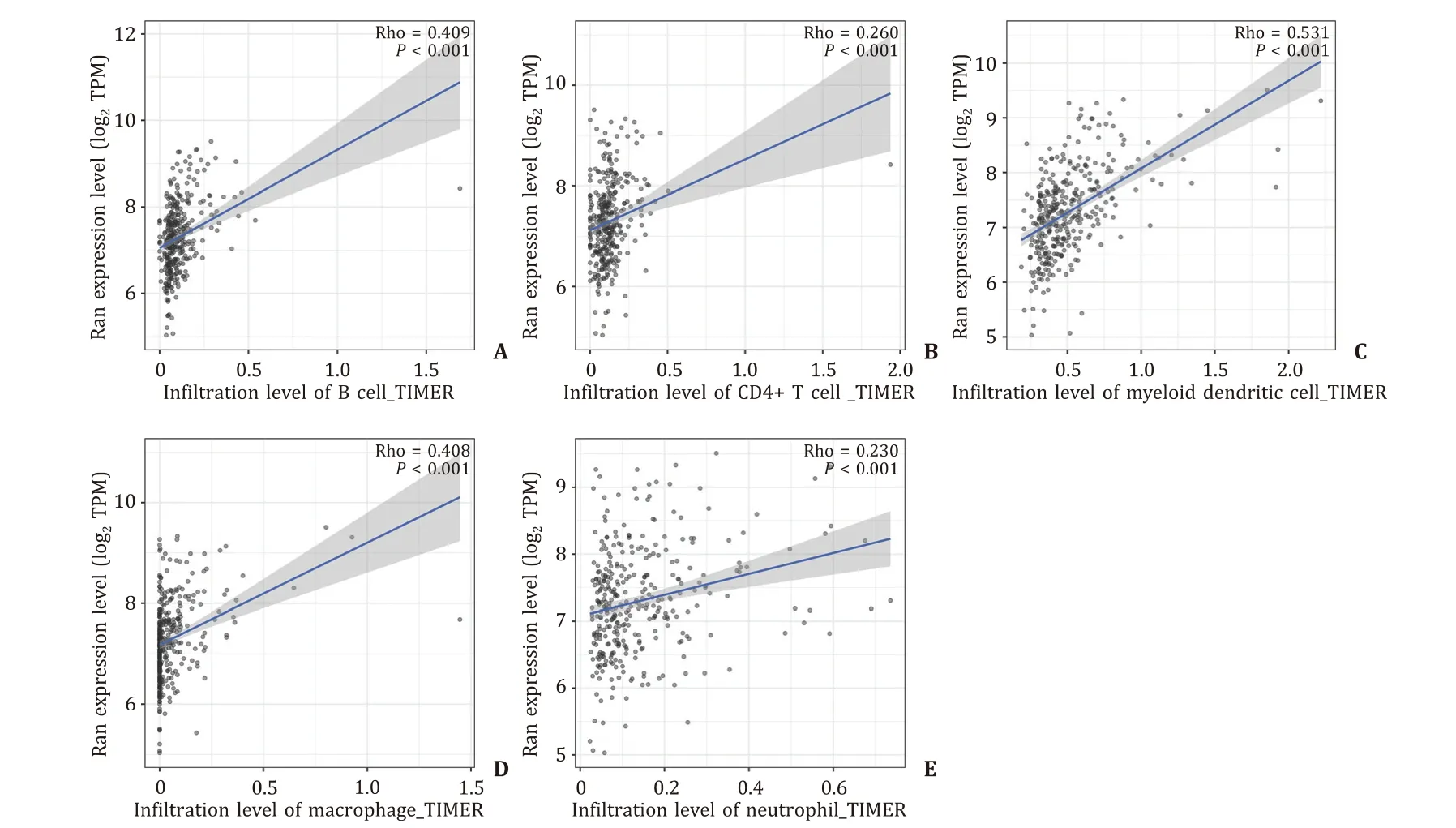
Fig. 5. Relationships of Ran expression with immune cells. A : B cells; B : CD4 + T cells; C : myeloid dendritic cells; D : macrophages; E : neutrophils.
Discussion
To the best of our knowledge, this is the first study to evaluate the role of Ran in predicting the prognosis of HCC patients.We found that Ran mRNA expression and promoter methylation were valuable prognostic factors in HCC patients. Importantly, our results revealed a negative correlation between Ran mRNA expression and promoter methylation in HCC. Ran expression level was correlated with grade, T stage, TNM stage and Ran methylation.However, Ran methylation was only associated with Ran mRNA expression. Additionally, we revealed that some immune cells and prognostic immune-related genes were related to Ran mRNA expression in HCC. High Ran expression indicated poor outcomes for HCC patients. The results of our study is consistent with those in other malignancies [ 14 , 16 , 20 ].
It is well-known that Ran expression was upregulated in colorectal cancer, breast cancer and head and neck squamous cell carcinoma tissues, compared with adjacent nontumor tissues. The overexpression of Ran indicated poor prognosis in these types of cancers [ 14 , 16 , 20 ]. With the restraint of growth factor regulation,Ran was activated in response to heregulin, a soluble secreted growth factor, which affected gene expression at mRNA processing level [21] . Ran is demonstrated to be involved in the process of cancer initiation and/or progression, such as proliferative signaling, resistance to cell death and activation of invasion and metastasis [22] .
Previous studies have shown that the development and progression of HCC are related to abnormal DNA methylation, thus,the detection of specific gene methylation could be used as a biomarker in early diagnosis, treatment targets and prognostic evaluation [ 23 , 24 ]. For instance, aberrant DNA methylation precisely delineated the transition from normal liver tissue to early HCC, and the DNA methylation pattern of the cirrhotic tumoradjacent tissue was selected as a survival predictor in HCC. Compared with nonmalignant tissue, HCC specimens obtained the highest heterogeneity in DNA methylation [23] . Since promoters of a wide range of tumor suppressor genes were epigenetically inhibited in HCC, the removal of epigenetic repression of those genes could reactivate their expression [24] . Given the dysregulated expression of Ran in HCC, we hypothesized that abnormal methylation of Ran might influence its expression. Focusing on promoter region, we found the inverse relationship between DNA methylation and mRNA expression for Ran. We also found that those hypermethylated Ran CpG sites were associated with longer OS and PFS.
There has been increasing evidence that tumor-infiltrating immune cells are involved in the development and progression of cancer [ 25 , 26 , 27 ]. In addition, immunotherapy targeting tumor microenvironment and immune infiltration has recently achieved satisfying effect in treating malignant tumors [26] . In a prior study,Ma et al. found that the expression of Aurora kinase A (Aurora-A) and ninein-interacting protein (AUNIP) showed a positive correlation with the infiltration of immune cells in HCC but a negative correlation with the infiltration of immune cells in lung adenocarcinoma [28] . A heterogeneity of immune infiltrate has been observed in primary colorectal cancer and synchronous and metachronous metastases. Moreover, high immune cell infiltration showed a strong association with prolonged patient survival [29] .It has been reported that the expression of HtrA serine peptidase 3(HTRA3) shows a negative correlation with the abundance of adaptive immune cells and a positive correlation with the abundances of innate immune cells [30] . Here we found that Ran expression was positively correlated with B cells, CD4 + T cells, myeloid dendritic cells, macrophages and neutrophils.
Although these findings contribute to the identification of a prognosis biomarker of HCC, there are still several limitations.To investigate the prognostic potential of Ran in HCC, we should take into consideration the different treatment methods received by patients. Nevertheless, the relevant information was absent in the TCGA database. Additional study regarding Ran methylation is needed to validate our results. Furthermore, Ran promoter methylation levels in HCC tissues and paired adjacent nontumor tissues require further investigation. Finally, aberrant Ran expression and its promoter methylation were found to be prognostic indicators in HCC in our study, but little is known about the underlying mechanism driving this process; thus, further study remains to be conducted.

Fig. 6. Correlations between Ran expression and prognostic immune-related genes.
In summary, Ran, which is negatively regulated by its methylation, possesses the potential to predict the outcome of HCC patients and may be involved in B cells, CD4 + T cells, myeloid dendritic cells, macrophages and neutrophils.
Acknowledgments
None.
CRediT authorship contribution statement
Hui-Hui Liu: Data curation, Formal analysis, Writing – original draft. Ju Wang: Methodology, Writing – original draft. Ying Zhang: Methodology, Writing – original draft. Yu-Chen Fan: Conceptualization, Funding acquisition, Supervision, Writing – review& editing. Kai Wang: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The study was supported by grants from the Key Project of the Chinese Ministry of Science and Technology ( 2017ZX10202202 and 2018ZX10302206 ), National Natural Science Foundation of China( 81970522 ), the Key Research and Development Project of Shandong Province ( 2019GSF108023 ), and Shandong University Multidisciplinary Research and Innovation Team of Young Scholars( 2020QNQT11 ).
Ethical approval
This study was approved by the Ethics Committee of Qilu Hospital of Shandong University (No. 2018 Scientific research express 515). Written informed consent was obtained from all participants.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Surgical exploration with non-resection in the setting of resectable,borderline and locally advanced pancreatic cancer
- Risk factors for post-endoscopic retrograde cholangiopancreatography(ERCP) abdominal pain in patients without post-ERCP pancreatitis
- On-table hepatopancreatobiliary surgical consults for difficult cholecystectomies: A 7-year audit
- Preoperative lymphocyte to C-reactive protein ratio as a new prognostic indicator in patients with resectable gallbladder cancer
- Relief of jaundice in malignant biliary obstruction: When should we consider endoscopic ultrasonography-guided hepaticogastrostomy as an option?
- Hepatobiliary&Pancreatic Diseases International