基于平行人工膜渗透性分析法初步预测盐酸吡格列酮制剂生物等效性
2022-06-01张丹丹李玉杰张冬梅于明艳杨蕙如陈德俊
张丹丹,王 松,李玉杰,张冬梅,于明艳,杨蕙如,陈德俊
(山东省食品药品检验研究院,国家药品监督管理局仿制药研究与评价重点实验室,山东省仿制药一致性评价工程技术研究中心,山东 济南 250101)
盐酸吡格列酮(pioglitazone hydrochloride)是一种噻唑烷二酮类胰岛素增敏剂,最早由日本武田药品工业株式会社(Takeda)成功合成,临床主要用于治疗2型糖尿病[1]。1999年7月,盐酸吡格列酮片获FDA批准上市,商品名为ACTOS(艾可拓)。日本药品审评中心(PMDA)公开的《IF文件》中显示:20 ℃下,盐酸吡格列酮在pH 1.1,pH 2.0,pH 5.0,pH 7.0溶液中的溶解度分别为6.7,0.42 ,0.00026,0.000093 mg/ml,因此,盐酸吡格列酮属于pH依赖性药物。在生物药剂学分类系统(biopharmaceutics classification system, BCS)中,盐酸吡格列酮属于II类,即低溶解性、高渗透性药物[2]。
口服固体制剂在人体内胃肠道的吸收取决于其溶出行为、生理条件下药物的溶解度和药物的渗透性。普通的溶出度实验可用于体外模拟胃肠道的崩解和溶出,但其反映的药物溶出行为变化不具临床意义[3],而人体生物等效性实验成本高,耗时长,失败风险大。测定药物渗透性最直接的方法是人体小肠灌流法,但是人体试验成本高且操作复杂。Caco-2细胞模型是国内外普遍采用的药物渗透性研究工具,Caco-2细胞来源于人的直肠癌,结构和功能与人小肠上皮细胞类似,广泛应用于药物转运机制、药物吸收等方面的研究,缺点是细胞培养周期长且易受污染[4]。动物模型也是研究药物渗透性的常用方法之一,主要包括体内小肠灌流试验和原位小肠灌流试验。体内小肠灌流试验更接近体内真实渗透情况。原位小肠灌流试验则模型相对简单,影响因素较少,参数易于分析。但是相关药物渗透性试验主要需用动物,试验成本高,时间长,需要考虑伦理学方面的问题,还可能出现假阴性的结果[4]。平行人工膜渗透性分析法(parallel artificial membrane permeability assay,PAMPA)是采用涂卵磷脂溶液的聚偏氟乙烯或聚碳酸酯膜模拟小肠上皮细胞以测定被动转运机制吸收的药物渗透性,其测定结果与人体小肠灌流法呈正相关[5-6]。采用PAMPA研究药物渗透性具有操作简便、效率高、重现性好等特点,且绝大多数口服固体制剂是通过被动转运机制渗透进入血液[7],因此,其在药物处方筛选及仿制药一致性评价等方面应用较广泛。
本实验将溶出度仪与人工仿生膜渗透性预测系统结合,建立了基于体外溶出-渗透速率测试结果的模型,具有一定的体外溶出与体内吸收相关性,用于预测盐酸吡格列酮片(胶囊)的生物等效性,并初步考察了辅料对盐酸吡格列酮原料渗透性的影响。受试制剂和参比制剂说明书中均表示空腹与餐后原形药物的药代动力学参数无明显差异,本次实验以空腹为代表。
1 材料与仪器
1.1 仪器
MacroFLUXTM型光纤药物溶出度与渗透速率测试系统(美国Pion Inc公司);MS105DU型电子天平与S400-K pH计(Mettler Toledo公司)。
1.2 药品与试剂
盐酸吡格列酮对照品(中国食品药品检定研究院,批号:100634-202104,纯度:100 %);参比制剂(盐酸吡格列酮片,Teva Takeda,商品名:艾可拓,规格:15 mg,批号:11887977);受试制剂A(盐酸吡格列酮片,规格:30 mg,已通过一致性评价),受试制剂B(盐酸吡格列酮片,规格:15 mg),受试制剂C(盐酸吡格列酮胶囊,规格:15 mg),3批受试制剂均来自山东省2021年风险监测抽验;渗透速率接收池(带膜)(美国Pion Inc公司,批号:520926);磷脂溶液(美国Pion Inc公司,批号:520852);ASB(pH 7.4)溶液(美国Pion Inc公司,批号:520945);药物溶出模拟液固体粉末(英国Biorelevant公司,批号:FFF-0421-B);氢氧化钠、氯化钠、无水磷酸二氢钠、盐酸、冰醋酸、二甲基亚砜(DMSO)均为分析纯;实验用水为纯化水。
2 方法与结果
2.1 MacroFLUXTM药物溶出度与渗透速率测试系统
本系统(见图1)中溶出杯作为供体室代表胃肠道,以不同pH值的溶出介质(空腹:胃液pH 1.6,肠液pH 6.5;饱腹:胃液肠液pH 5.0)进行空饱腹实验(本实验以空腹为代表),用涂以磷脂溶液的人工仿生膜模拟胃肠道细胞膜,受体室中加入ASB缓冲液(pH 7.4)用于模拟血液。制剂置于溶出杯中模拟药物的溶出行为,溶解后的药物通过仿生膜进入受体室模拟药物的吸收过程。药物浓度变化通过原位光纤DAD检测器实时检测,实现药物溶出和渗透吸收同时监测。通过关键质量参数考察药物的释放与吸收过程,预测制剂间是否生物等效。

图1 MacroFLUXTM药物溶出度与渗透速率测试系统示意图
2.2 标准曲线的建立
2.2.1 对照品储备液的配制 取盐酸吡格列酮对照品适量,精密称定,用DMSO制成每1 ml含吡格列酮8.1,1.0 mg两种浓度的溶液,分别作为供体室对照品储备液和受体室对照品储备液。
2.2.2 供体室标准曲线的建立 胃模拟液(pH 1.6):将光纤探头置入200 ml胃模拟液(pH 1.6),每次加入90 μl的供体室对照品储备液后测定,得每1 ml含吡格列酮3.7,7.3,11.0,14.7,18.3 μg的一系列溶液的紫外光谱,选取286~296 nm波长范围,以吸光度二阶导数为横坐标,以浓度为纵坐标进行线性回归,建立盐酸吡格列酮制剂(规格:15 mg)胃模拟液(pH 1.6)中的标准曲线。同法操作,每次加入180 μl的供体室对照品储备液后测定,建立盐酸吡格列酮制剂(规格:30 mg)胃模拟液(pH 1.6)中的标准曲线。
空腹肠模拟液(pH 6.5):将光纤探头置入200 ml空腹肠模拟液(pH 6.5),每次加入50 μl的供体室对照品储备液后测定,得每1 ml含吡格列酮2.0,4.1,6.1,8.2,10.2 μg的一系列溶液的紫外光谱,照胃模拟液(pH 1.6)下方法处理,建立盐酸吡格列酮制剂(规格:15,30 mg)空腹肠模拟液(pH 6.5)中的标准曲线。
以上标准曲线的线性相关系数均大于0.9999,线性关系良好。
2.2.3 受体室标准曲线的建立 将光纤探头置入50 ml ASB(pH 7.4)溶液中,每次加入50 μl的受体室对照品储备液后测定,得每1 ml含吡格列酮1.0,2.1,3.1,4.1,5.2 μg的一系列溶液的紫外光谱,按照胃模拟液(pH 1.6)中的方法处理后建立受体室标准曲线。此标准曲线的线性相关系数大于0.9999,线性关系良好。
2.3 测定方法
采用图1装置,按溶出度与释放度测定法(《中国药典》2020年版四部通则0931第二法),溶出介质:开始以胃模拟液(pH 1.6)800 ml为溶出介质,30 min时加入混合溶液[空腹肠模拟液(10×)与磷酸盐缓冲液等比混合]200 ml使成空腹肠模拟液(pH 6.5);温度:37.0 ℃;转速:50 r/min;受体室(即渗透速率接收池)接收液:ASB缓冲液(pH 7.4)12 ml;微搅拌桨转速:450 r/min;以50 μl胃肠道模拟脂质体溶液浸润渗透速率接收池上的仿生膜(膜面积3.88 cm2),采集时间300 min,采样间隔1 min。在240~400 nm内,用光纤探头分别测定溶出杯和受体室中的吸光度,按公式(1)、(2)分别计算制剂的渗透速率J(FLUX)和总渗透量T(permeation amount)。采用双侧t检验的方法,按照下式(3)计算渗透速率和总渗透量几何均值比的90%置信区间。

式中J为渗透速率,即药物单位时间通过单位膜面积的量[μg/(min·cm2)];为单位时间内受体室中药物浓度变化速率[μg/(ml·min)];V为受体室中接受液的体积(ml);A为仿生膜膜面积(cm2,本次实验为3.88 cm2)。

式中T为总渗透量(permeation amount),即药物在测定时间通过仿生膜的总量(μg);C为最终时间点接受液中的药物浓度(μg/ml);V为受体室中接受液的体积(ml)。

式中JT为受试制剂渗透速率的平均值,当计算总渗透量的90 %置信区间时,为受试制剂总渗透量的平均值;JR为参比制剂渗透速率的平均值,当计算总渗透量的90 %置信区间时,为参比制剂总渗透量的平均值;S为受试制剂渗透速率(或总渗透量)的标准偏差;n为样本重复次数(本次实验n=3);C为换算系数,与重复次数具有相关性,n=3时,C=2.92。
受试制剂A规格为30 mg,选取的参比制剂规格为15 mg,在进行受试制剂A的生物等效性试验时,参比制剂每溶出杯中投药2片。进行受试制剂B、C等效性实验时,受试制剂与参比制剂每溶出杯投药1片。
2.4 生物等效性预测结果
受试制剂A在30 min时溶出杯中浓度可达30 μg/ml(见图2 供体室),表明此时盐酸吡格列酮溶出完全。在加入混合溶液后溶出介质由胃模拟液(pH 1.6)转换为空腹肠模拟液(pH 6.5),溶出杯中盐酸吡格列酮逐渐析出,浓度降低并维持约2.5 μg/ml(见图2 供体室、图3供体室)。受体室中前30 min盐酸吡格列酮一直维持在极低浓度水平,表明药物渗透较少,溶媒转换至空腹肠模拟液(pH 6.5)后,受体室中吡格列酮浓度逐渐增加(见图2受体室、图3受体室)。30 min之前,盐酸吡格列酮在胃模拟液(pH 1.6)中快速溶解,药物浓度较高,但大部分呈解离状态以离子形式存在,难以跨膜渗透;在空腹肠模拟液(pH 6.5)中虽溶解度降低,但多以分子形式存在易于跨膜。本实验截取受体室120~170 min数据,根据受体室中测得的浓度,以时间对浓度进行线性回归,各受试制剂与参比制剂的线性方程见表1,线性相关系数均大于0.995。以该时间段药物浓度变化速率(即线性方程斜率)代入式(1)计算渗透速率,以300 min时测得的受体室接收液浓度(见表1)代入式(2)计算总渗透量,受试制剂A与参比制剂(投药2片)计算结果(n=3)见表2,受试制剂A渗透速率和总渗透量几何均值比的90 %置信区间在80.00 %~125.00 %之间,可初步预测其与参比制剂生物等效。鉴于受试制剂A已通过一致性评价,可证明此系统用于盐酸吡格列酮制剂生物等效性初步预测的可行性及准确性。

表1 渗透速率线性方程及最终点受体室接受液浓度
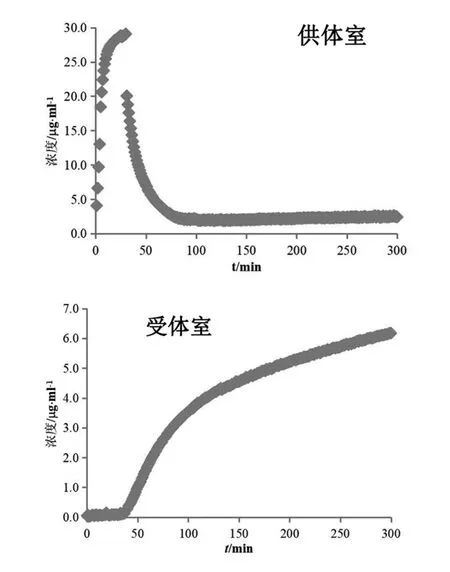
图2 受试制剂A溶出(供体室)与渗透(受体室)浓度变化图
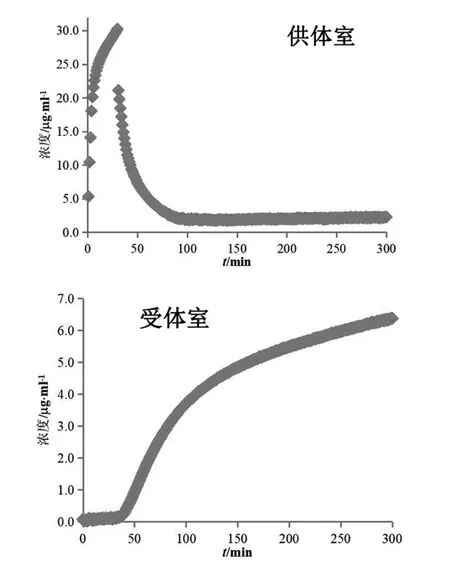
图3 参比制剂(投药2片)溶出(供体室)与渗透(受体室)浓度变化图

表2 受试制剂A与参比制剂(投药2片)渗透速率与总渗透量计算结果
对受试制剂B及受试制剂C进行生物等效性预测,计算结果(n=3)见表3。两制剂的渗透速率和总渗透量几何均值比的90 %置信区间均未落在80.00 %~125.00 %,可初步预测受试制剂B、C与参比制剂生物不等效。

图4 受试制剂B溶出(供体室)与渗透(受体室)浓度变化图
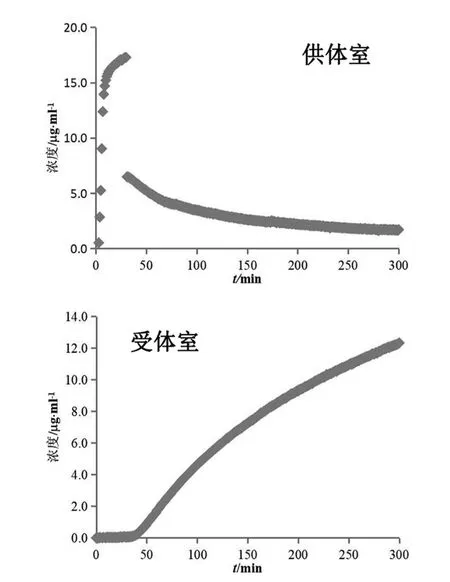
图5 受试制剂C溶出(供体室)与渗透(受体室)浓度变化图
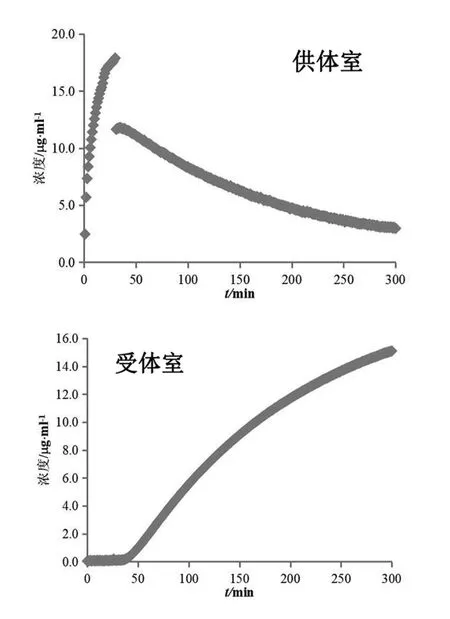
图6 参比制剂(投药1片)溶出(供体室)与渗透(受体室)浓度变化图
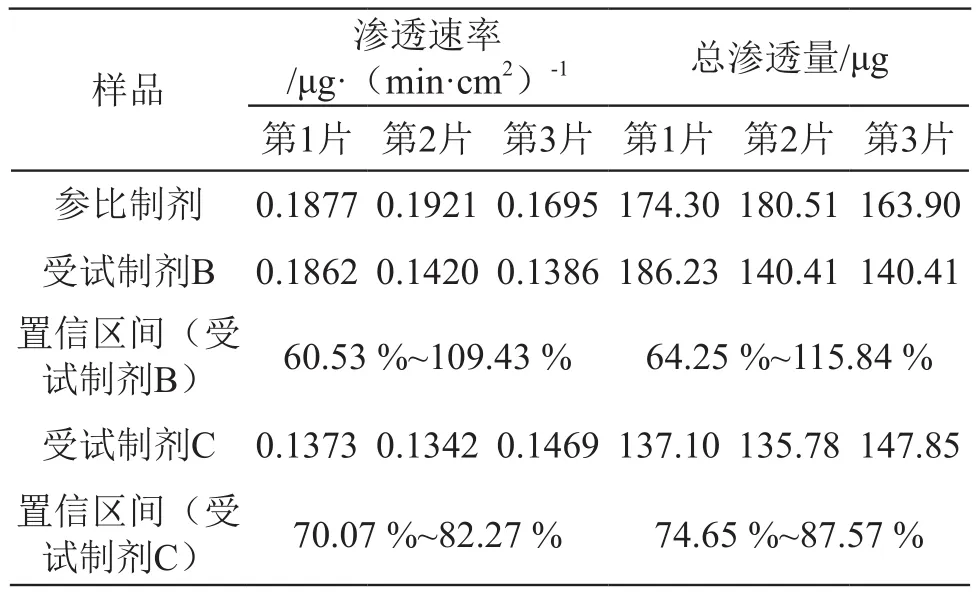
表3 受试制剂B、C与参比制剂(投药1片)渗透速率与渗透总量计算结果
2.5 辅料对吡格列酮渗透性影响
将3批受试制剂的原料(称取相当于其规格的重量)投入供体室中,通过与制剂渗透结果对比,以考察辅料对吡格列酮渗透性的影响。3批受试制剂的原料300 min时渗透浓度约20 μg/ml,受试制剂B和C 于300 min时渗透浓度约为11 μg/ml,而受试制剂A仅为6 μg/ml(见图7),由此可见,3批受试制剂原料的渗透总量和渗透速率均远高于其制剂。鉴于3批受试制剂在30 min内均能溶出完全,药物的渗透性不再受其溶出的限制,故而辅料(种类、用量等)成为影响主成分渗透性的关键因素。
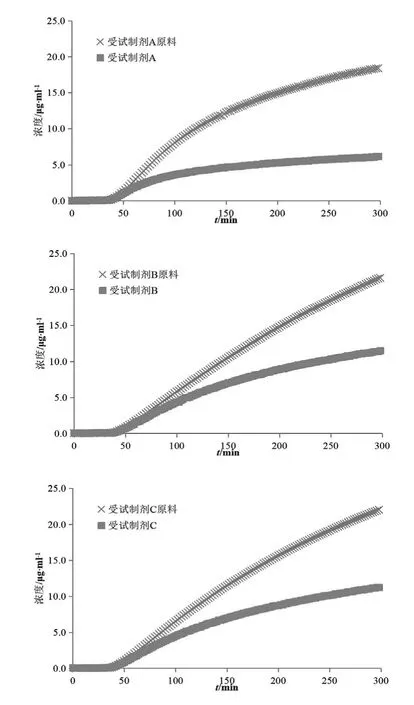
图7 受试制剂A、B、C与其原料渗透性测定结果对比图
3 讨论
盐酸吡格列酮是pH依赖型药物,在胃模拟液(pH 1.6)中溶解度良好,溶媒转换为空腹肠模拟液(pH 6.5)后因溶解度降低逐渐有沉淀析出(见图8),辅料的用量、种类都可能成为影响沉淀粒子大小的因素。有文献报道胃肠道中盐酸吡格列酮析出后的粒径影响药物吸收,进而导致生物利用度差异[8]。提示在类似pH依赖型药物的制剂处方筛选时因溶解度导致药物析出沉淀的粒径也值得关注。

图8 受试制剂A、B在胃模拟液(pH 1.6)及空腹肠模拟液(pH 6.5)中溶解情况
对比不同规格盐酸吡格列酮制剂的实验数据发现,15 mg规格的渗透速率和总渗透量均高于30 mg规格。参比制剂投药2片与投药1片供体室、受体室中吡格列酮浓度变化对比情况见图9。前30 min供体室介质为胃模拟液(pH 1.6),因吡格列酮能完全溶解,30 mg规格的供体室浓度远高于15 mg规格。半小时后溶媒转换为空腹肠模拟液(pH 6.5),因溶解度降低吡格列酮析出。由于30 mg规格过饱和浓度高,析出较15 mg规格快,自40 min开始供体室中浓度一直低于15 mg规格。通过对比原料(图10)与制剂(图9 供体室)在供体室的浓度变化发现,原料的过饱和析出较制剂缓慢。这说明吡格列酮在过饱和析出的过程中包裹辅料或者辅料的某些性质(用量、溶解性等)加速了这种析出。本预测系统是模拟基于浓度驱动的渗透行为,也就导致了30 mg规格的渗透低于15 mg规格。鉴于生物等效性研究以同等剂量的制剂相比较,所以此现象对本预测系统在生物等效性研究方面的应用影响较小。本实验采用的溶出-人工仿生膜渗透性预测系统直观地模拟了盐酸吡格列酮体内溶出渗透过程,为盐酸吡格列酮制剂的处方筛选和生物等效性初步预测提供了新思路和方法。
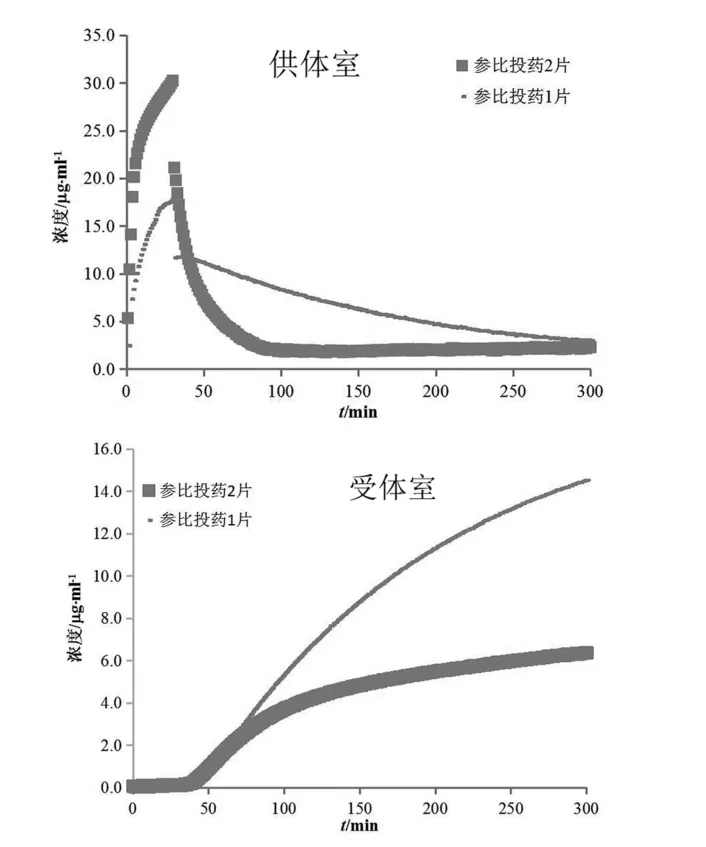
图9 参比制剂投药2片与投药1片溶出(供体室)、渗透(受体室)浓度对比图
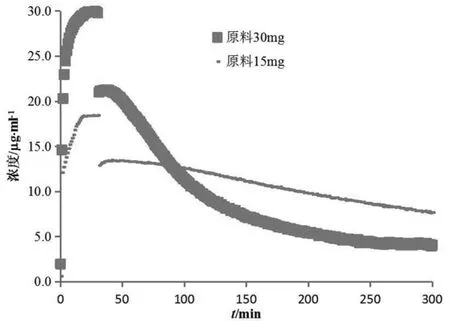
图10 原料30 mg与原料15 mg供体室浓度对比图
