Hippo-YAP信号通路调控细胞衰老的分子机制*
2022-05-31詹永通吴桂豪范旭红陈路苗陈同生王小平
詹永通, 吴桂豪, 范旭红, 陈路苗, 陈同生, 王小平△
(1暨南大学附属第一医院疼痛科,广东 广州 510630;2华南师范大学生物光子学研究院,广东 广州 510631)
作为Hippo 信号通路的主要效应蛋白,Yes 相关蛋白(Yes-associated protein,YAP)主要通过与转录增强相关结构域(transcriptional enhanced associate domain,TEAD)/非TEAD 转录因子结合的方式调控靶基因转录[1]。YAP 蛋白的结构主要包括WW 结构域(介导非TEAD 转录因子的结合)、TEAD 结合域(介导TEAD 转录因子的结合)、C 端转录激活域(介导转录激活)、C 末端PDZ 结合域(核定位所需)、卷曲螺旋域(介导蛋白质-蛋白质的相互作用)、N 末端富含脯氨酸基序(抑制转录活性)和SH3 结合基序(介导含SH3 结构域蛋白的相互作用)[2]。YAP 广泛参与细胞增殖与细胞分化的调控,是不同器官生长与再生所必需的转录共激活因子,在器官再生与再生医学领域被广泛研究[3]。同时YAP 通过调控细胞增殖以及凋亡,影响肿瘤的发生、发展与转移,是目前肿瘤相关研究的重点[4]。另外,近年来YAP 调控细胞衰老的研究也逐渐增多[5-7]。
细胞衰老是细胞应激因素诱导的细胞周期永久性停滞状态[8]。细胞衰老分为复制性衰老和压力诱导的过早衰老。端粒短缩、DNA 损伤、氧化应激、炎症、原癌基因激活等促衰老因素主要通过p53-p21-RB(retinoblastoma protein)和p16INK4A-RB 两种信号通路的激活来诱导细胞衰老。细胞发生衰老后具有永久性生长停滞、分泌衰老相关表型(senescence-associated secretory phenotype,SASP)及抵抗凋亡等特征。急性衰老细胞具有修复受损组织以及预防恶性肿瘤发生等积极作用[9-10]。而慢性衰老细胞会导致衰老细胞的积累以及衰老相关分泌表型的长期影响,最终导致一系列疾病的发生[11]。研究表明慢性细胞衰老与癌症[12]、动脉粥样硬化[13]、骨关节炎[14]等年龄相关疾病的发生发展相关。选择性清除衰老细胞可以延缓小鼠肿瘤的进展,改善动脉粥样硬化[13],减轻骨关节炎的疾病进展[15]以及延长小鼠健康寿命等作用[16]。Hippo-YAP 信号通路主要与组织发育、再生以及稳态等方面有关[1]。同时研究提示Hippo-YAP 信号通路与细胞衰老的发生存在一定的联系,可能是调控细胞衰老的新途径[5-7]。
本文介绍了Hippo-YAP 信号通路的结构及功能、Hippo-YAP 信号通路调控细胞衰老的分子机制以及Hippo-YAP 信号通路对衰老相关疾病的治疗现状的研究进展,期望为衰老相关疾病的分子机制研究及临床疾病治疗提供参考。
1 Hippo-YAP信号通路
作为Hippo信号通路的主要效应蛋白,YAP广泛参与肿瘤的发生与发展、细胞的增殖与分化以及细胞的凋亡与衰老的调控[17]。YAP 是一种转录共激活因子,需要与细胞核内的TEAD/非TEAD 转录因子结合后才可调节靶基因的表达。当Hippo 信号通路被激活,通过一系列激酶级联反应,YAP 发生磷酸化被滞留在细胞质,进而被蛋白酶水解而失活[3]。而当Hippo信号通路失活,未被磷酸化的YAP会被转移进细胞核与TEAD 转录因子结合进而发挥其多种生物学作用。YAP 作为协调生物力学信号、能量代谢及发育信号级联之间的纽带,在细胞生长发育中发挥重要的作用[18]。
1.1 YAP 激活过程 YAP 的激活过程包括经典的Hippo 信号通路以及非依赖Hippo 信号通路两种途径。生物力学信号,可溶性配体,应激以及组织损伤与炎症等上游激活因素可通过经典Hippo 信号通路调节YAP 的活性[19-22]。上述激活因素可引起哺乳动物不育系20 样激酶1/2(mammalian sterile 20-like kinase 1/2,Mst1/2)发生磷酸化激活,然后引起萨尔瓦多家族同源物1(Salvador homolog 1,SAV1)及MOB(Mps one binder)1A/B 的磷酸化[23]。磷酸化的SAV1与磷酸化的Mst1/2 形成复合体后可招募大肿瘤抑制因子1/2(large tumor suppressor 1/2,LATS1/2)并使其磷酸化而激活。激活后的LATS1/2 与前面被激活的MOB1A/B 形成的蛋白聚合物可导致YAP 发生磷酸化(图1)[18]。其中MST1/2 和LATS1/2 的磷酸化是经典Hippo 信号通路的关键步骤。部分上游调节因素也可以直接引起LATS1/2 磷酸化。磷酸化的YAP 与14-3-3 蛋白结合后滞留在细胞质中,被酪蛋白激酶1δ/1ε 进一步磷酸化和SCFβ-TRCPE3 泛素连接酶复合物泛素化,最后被蛋白酶体降解[24](图1)。若Hippo信号通路被抑制,不被磷酸化的YAP 会被转移进细胞核而激活。除了经典的Hippo信号通路,上游激活因素也可以通过非依赖Hippo 信号通路激活YAP。非依赖Hippo 信号通路不依赖于MST1/2 及LATS1/2等关键酶的激活,而是通过肌动蛋白的张力传导改变细胞核孔的渗透性或者其他蛋白直接磷酸化YAP来调控YAP 的活性。目前研究表明,黏着斑(focal adhesion,FA)、黏附连接(adherens junctions,AJs)、紧密连接(tight junctions,TJs)、Wnt-β-连环蛋白、甲基化与磷酸化、Notch 信号通路及TGF-β 信号通路可以不依赖Hippo信号通路调节YAP的激活[25-27]。

Figure 1. Activation of Hippo-Yes-associated protein (YAP)signaling pathway. MST1/2:mammalian sterile 20-like kinase 1/2;SAV1:Salvador homolog 1;MOB1A/B:Mps one binder 1A/B;LATS1/2:large tumor suppressor 1/2;P:phosphorylation.图1 Hippo-YAP信号通路激活过程
1.2 参与Hippo-YAP 信号通路调节的上游激活因素
1.2.1 生物力学信号 细胞外基质硬度、细胞密度及流体剪切力三部分生物力学信号通过机械转导作用,将细胞外坏境的机械信号转换为生物化学信号,引起细胞YAP 活性的改变,进而促进细胞适应外坏境的变化[28-29]。
细胞外基质主要通过改变LATS1/2 活性及细胞核孔通透性来调节YAP 的活性。一方面,细胞外基质的硬度可以作用于细胞表面的FA 和其他黏附蛋白,影响应力纤维细胞骨架的张力状态,通过一系列酶的激活,引起LATS1/2 活性的改变,进而最终影响YAP 的活性。根据细胞类型的不同可分为两种途径:整合素(integrin)-FA 激酶(FA kinase,FAK)-SRC(steroid receptor coactivator)-PI3K(phosphatidylinositol 3-kinase)-PDK1(3-phosphoinositide-dependent protein kinase 1)[30]和 整 合 素-FAK-p130Cas-Rac1-PAK(p21-activated kinase)-merlin[31]。另一方面,FA 感受到细胞外基质硬度改变后,会引起应力纤维细胞骨架的张力状态发生改变,通过细胞核骨架和细胞骨架的连接子(linker of nucleoskeleton and cytoskeleton,LINC)来调节YAP 活性。当细胞在硬的细胞外基质中,应力纤维和LINC 形成的粘连将机械力传递到细胞核,导致细胞核的拉伸,进而增加核孔的渗透性,引起YAP进入细胞核增多而激活[25];当细胞在软的细胞外基质中,应力纤维与LINC 解偶联,无法通过LINC 将机械力转递给细胞核,细胞核孔不发生改变,YAP滞留在细胞质而被降解[32]。
细胞密度主要通过FA、AJs 及TJs 三种途径来改变YAP 活性。细胞密度的FA 途径与细胞外基质硬度的调节相似。当细胞密度降低时,每个细胞表面的FA 数量增加,引起连接到FA 上的F-肌动蛋白细胞骨架的张力增加,LATS1/2活性或者细胞核孔大小发生改变[33]。细胞密度的AJs 途径主要通过α-连环蛋白结合merlin 蛋白来实现的。当细胞密度增加,α-连环蛋白作为AJs 的成分之一随之增加,结合的merlin 蛋白增加;merlin 蛋白可以结合并隔离LATS1/2,但是会引起LATS1/2 反馈性增加,引起YAP 磷酸化增加,YAP 活性降低,细胞增殖受到抑制[34]。细胞密度的TJs 途径主要通过血管动蛋白样蛋白2(angiomotin-like protein 2,AMOTL2)介导。AMOTL2可以直接激活LATS2并促进LATS2与YAP结合。当细胞密度增加,AMOTL2 数量增加,活化的LATS2 增加,磷酸化的YAP增加,细胞增殖同样受到抑制[35]。
流体剪切力同样可以改变YAP的活性。研究表明,层流液体所致的剪切力会抑制YAP活性,而湍流液体所致的剪切力会促进YAP进入细胞核。同时流体剪切力也可以影响间充质干细胞和软骨细胞的YAP 表达水平。研究表明,流体剪切力主要是通过整合素、Gα13和Rho起作用的[36]。
1.2.2 可溶性配体 可溶性配体(细胞外配体)通过与对应的细胞表面受体结合,引起MST1/2 或LATS1/2 活性改变来调节YAP 的磷酸化。根据细胞表面受体的不同,主要分为两类:G 蛋白偶联受体(G-protein-coupled receptor,GPCR)可溶性配体和受体酪氨酸激酶(receptor tyrosine kinase,RTK)可溶性配体。一方面,溶血磷脂酸(lysophosphatidic acid,LPA)、鞘 氨 醇-1- 磷 酸(sphingosine-1-phosphate,S1P)、胰高血糖素和肾上腺素等配体与GPCR 结合后,激活异源三聚体G12/13、Gq/11 和Gi/o,进而抑制LATS1/2 磷酸化,促进YAP 转移进细胞核增加[20]。上述配体也可以激活异源三聚体Gs,引起LATS1/2磷酸化增加,YAP 活性降低[20]。另一方面,表皮生长因子(epidermal growth factor,EGF)结合RTK 后通过PI3K-PDK1 信号通路抑制MST1/2 的磷酸化作用,进而使LATS1/2活化,促进YAP转移进细胞核增加[30]。
1.2.3 能量应激、渗透压应激和缺氧应激 能量应激、渗透压应激及缺氧应激等细胞微坏境压力也将影响YAP的活性。在营养缺乏和ATP抑制剂等能量应激的情况下,胞质中的AMP 活化蛋白激酶(AMPactivated protein kinase,AMPK)活 化,活 化 后 的AMPK 一方面可以引起LATS1/2 激活而增加YAP 的磷酸化,另一方面可以直接磷酸化YAP 的第94 位丝氨酸残基,阻碍YAP 与TEAD 转录因子结合而抑制YAP 功 能[37]。渗 透 压 应 激 通 过Nemo 样 激 酶(Nemo-like kinase,NLK)引起YAP 第128 位丝氨酸发生磷酸化,从而抑制14-3-3 与YAP 的结合并促进YAP 转移进细胞核[38]。14-3-3 通过结合YAP 而促进YAP 在胞质降解,所以抑制14-3-3 与YAP 的结合也促进了YAP 的细胞核转移[38]。此外,缺氧应激则通过SIAH2 泛素E3 连接酶抑制LAST2 的功能来促进YAP的核转移[39]。
1.2.4 炎症和组织损伤 肝、皮肤、心肌及肠上皮的损伤会引起活化的YAP 增加,进而促进损伤组织的修复[40]。同时炎症细胞因子,如前列腺素E2和肿瘤坏死因子也可激活YAP[22]。目前炎症及组织损伤如何调控YAP的活性尚不清楚,有待进一步研究。
1.3 YAP 结合的转录因子 YAP 作为转录共激活蛋白,需与转录因子结合后才能发挥其生物学功能。根据转录因子是否含有TEAD 分为TEAD 转录因子和非TEAD 转录因子。TEAD 转录因子在哺乳动物中可分为4 型:TEAD1、TEAD2、TEAD3 和TEAD4,几乎在所有类型的组织中均有表达[41]。TEAD 转录因子的结构包括N 端的DNA 结合域(DNA binding domain,DBD)、C 端的YAP 结合域(YAP binding domain,YBD)及 富 含 脯 氨 酸 的 氨 基 酸 序 列[42]。TEAD1~4 具有61%~73%的整体同源性,并且DBD及YBD 具有90%以上的同源性[41]。TEAD1 与心肌细胞分化发育的关系较大,TEAD2 可能会影响神经发育相关基因的表达,TEAD3 主要功能的相关研究仍较少,TEAD4 主要影响胚胎植入相关基因的表达[43]。YAP 与TEAD1~4 转录因子结合后,影响下游基因表达,对细胞的增殖、分化、凋亡及衰老等细胞行为进行调控。YAP 结合的非TEAD 转录因子主要包 括Smad2/3、RUNX2/3、p63、p73、octamer-binding transcription factor 4 (OCT4)、PR/SET domain 4(PRDM4)、MYC、Krüppel-like factor 5(KLF5)、Erb-B2 receptor tyrosine kinase 4(ERBB4)和early growth response-1(EGR-1),并且上述非TEAD 转录因子主要与肿瘤发生及转移有关[44]。
1.4 YAP的主要生物学作用
1.4.1 YAP 参与调控肿瘤发生与发展 研究表明,除了血液系统恶性肿瘤和睾丸癌以外,YAP 在肺癌、乳腺癌、肝细胞癌、子宫颈鳞状细胞癌、黑色素瘤等大部分肿瘤中被异常激活[45]。YAP 被异常激活后,通过与p63、MYC 等促肿瘤相关转录因子结合,影响细胞周期和细胞分裂相关蛋白的表达水平,诱导细胞的强烈增殖,导致肿瘤的发生[46](图2)。在肺癌中,YAP 的高表达或活性增高与肺癌的高恶性程度和不良预后存在联系[47]。在乳腺癌中,条件性敲除YAP可以减少小鼠乳腺癌的生长[48],而YAP的过表达则可以赋予良性乳腺肿瘤肺转移的能力[49]。在肝细胞癌中,敲减YAP或TAZ有效抑制人和小鼠肝细胞癌细胞系的皮下肿瘤生长[50],而YAP过表达则导致非致瘤性人肝细胞系出现致瘤性[46]。在膀胱癌中,使用敲减YAP表达水平可降低膀胱癌细胞活力并促进其凋亡[51]。但是在头颈癌的研究中显示,YAP 主要发挥抑制肿瘤发生的作用,并且敲减YAP增强了头颈癌细胞系的增殖、存活、迁移和对顺铂的抗性[52]。在早幼粒细胞白血病中,YAP-1 作为一种肿瘤抑制因子诱导肿瘤细胞发生凋亡[53]。目前YAP在肿瘤中的作用仍存在争议,不同肿瘤类型中YAP作用的差异可能与组织和细胞环境特异性有关[54-55]。
1.4.2 YAP 参与调控细胞增殖与分化 YAP 在体内外可以促进不同类型的细胞增殖。一方面,YAP与TEAD 转录因子结合可以直接影响有丝分裂激酶、DNA 复制蛋白及DNA 修复酶等细胞周期相关蛋白的表达水平,维持细胞的增殖能力[56-57](图2);另一方面,YAP 通过增加下游基因(如MYC等)的表达间接影响细胞增殖[58]。同时YAP 可以上调BCL-2(B-cell leukemia/lymphoma-2)、MCL-1(myeloid cell leukemia-1)等抗凋亡基因的表达,来维持细胞的存活[59](图2)。在小鼠体内模型敲减MST1和MST1/2,或过表达YAP可以驱动肝细胞和心肌细胞的增殖,使肝脏和心脏过度生长。除了驱动细胞增殖,YAP还可以促进细胞去分化以及获得干细胞特性。研究表明YAP 在各种干细胞中高表达,而在分化细胞中低表达[60]。在体外,通过基因编辑手段过度激活YAP可以诱导细胞去分化并且重新成为干细胞[3]。
1.4.3 YAP 参与调控细胞凋亡与衰老 YAP 除了参与调控肿瘤发生和细胞增殖外,还参与调控细胞凋亡与细胞衰老。在调控细胞凋亡方面,YAP 通过影响抗凋亡和促凋亡基因的表达水平来发挥抗凋亡作用和促凋亡作用,这种作用差异主要取决于YAP结合的转录因子类型[55]。一方面,YAP 与TEAD 转录因子结合,促进BCL-2、MCL-1、环加氧酶2(cyclooxygenase-2,COX-2)、存活蛋白(survivin)及葡萄糖转运蛋白1(glucose transporter 1,Glut1)等抗凋亡基因的表达来抵抗凋亡的发生[61-62](图2);另一方面,YAP 通过与非TEAD 转录因子p73 结合,促进BAX(BCL-2-associated X protein)、PUMA(p53-upregulated modulator of apoptosis)等促凋亡基因的表达来发挥促凋亡作用[53,63](图2)。在调控细胞衰老方面,YAP 通过影响p53、p16和CDK6(cyclin-dependent kinase 6)等下游基因的表达水平或者细胞的自噬水平等途径调控细胞衰老的发生(图2)。在IMR90 细胞中,YAP 通过依赖TEAD 转录因子、p53、p16 和CDK6的方式抑制细胞衰老的发生,其中CDK6是YAP 调控细胞衰老的直接下游靶基因[64]。在HCT116 细胞中,Werner 综合征蛋白(Werner syndrome protein,WRN)的缺失会引起YAP 的激活,从而加速细胞衰老的发生[65]。在星形胶质细胞中,YAP 通过促进CDK6 蛋白的表达水平来抑制星形胶质细胞的衰老[66]。在内皮细胞中,YAP 通过阻断自噬流和激活哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路促进内皮细胞和血管组织的衰老[6]。因此,YAP 调控细胞衰老的作用与细胞类型相关[6,64-67]。
2 Hippo-YAP信号通路调控细胞衰老的机制
2.1 YAP-ATM(ataxia-telangiectasia mutated)-p53-p21 ATM-p53-p21 是细胞衰老发生的主要信号通路之一。细胞复制过程中或者氧化应激等条件下,会引起DNA 的损伤以及随后的DNA 损伤应答(DNA damage response,DDR)。DDR 会激活ATM 激酶,随后ATM 激酶通过依赖细胞周期检测点激酶2(cell cycle checkpoint kinase 2,CHK2)的方式磷酸化p53。磷酸化的p53 可以避免MDM2(mouse double minute 2)介导的蛋白酶体降解,并且激活下游靶基因CDKN1A(cyclin-dependent kinase inhibitor 1A),引起p21 的表达水平增加[68]。p21 作为CDK 抑制剂,最终引起了细胞周期的停滞[69](图3)。Hippo信号通路核心成分与p53 之间存在复杂的相互作用关系,共同控制干细胞发育、细胞凋亡、细胞衰老等过程。在细胞衰老方面,Hippo信号通路核心成分LATS2通过结合并抑制MDM2功能,从而稳定p53不被降解并激活p53 的功能,诱导细胞衰老的发生[70](图3)。并且p53 作为转录因子作用下游靶基因引起LATS2 的表达增加,从而在p53 与LATS2 之间形成一种正反馈调节,进一步促进细胞衰老的发生[71]。Hippo信号通路的主要效应蛋白YAP 同样与p53 之间存在相互作用并调控细胞衰老的发生。研究表明,胞质中的p53凋亡刺激蛋白1(apoptosis-stimulating protein of p53 1,ASPP1)可以结合并抑制LATS1的功能,引起YAP核积累增加。核内的YAP 与转录因子结合后导致LATS2 表达降低,从而降低了p53 诱导的p21 表达,阻止细胞周期停滞和细胞衰老发生[72](图3)。同时,研究表明YAP 与p73 转录因子结合后促进PML(promyelocytic leukemia protein)的表达,PML 蛋白与YAP 之间形成的复合物可以保护YAP 免于降解,并且该复合物促进了p53 的活化,最终促进细胞衰老的发生[65]。另外,YAP 作为转录共激活因子,可以直接影响p53基因的表达水平,从而影响p53 的功能[73](图3)。

Figure 2. Yes-associated protein-transcription factors(YAP-TF)regulate cell proliferation,differentiation,apoptosis,senescence and tumorigenesis and development. BCL-2:B-cell leukemia/lymphoma-2;BAX:BCL-2-associated X protein;PUMA:p53-upregulated modulator of apoptosis;MCL-1:myeloid cell leukemia-1;Pol II:polymerase II;CDK9:cyclin-dependent protein kinase 9;AP-1:activating protein-1;SOX2:sex-determining region Y-box 2;OCT4:octamer-binding transcription factor 4.图2 YAP-转录因子调控细胞增殖、分化、凋亡、衰老及肿瘤发生发展
2.2 YAP-p16INK4A-CDK-RB p16INK4A-CDK-RB 是细胞衰老发生的另一主要信号通路。DNA 损伤、氧化应激等促衰老因素分别通过p38 丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)[74]和细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)控制B 细胞特异性Moloney 鼠白血病病毒插入位点1(B-cell-specific Moloney murine leukemia virus insertion site 1,BMI1)和转录因子ETS1/2,引起p16INK4A表达增加[75]。随后p16INK4A抑制CDK4 和CDK6 磷酸化RB 的能力,使RB 维持在低磷酸化状态。低磷酸化的RB 结合和隔离转录因子E2F、DP1及DP2,抑制S 期基因的表达,引起细胞周期停滞[68](图3)。IMR90 成纤维细胞的研究表明,敲减YAP通过TEAD 和p53/p16/CDK6 依赖的方式促进细胞衰老,并且CDK6作为YAP-TEAD 转录因子的下游靶基因参与细胞衰老的调控[64](图3)。YAP缺乏导致的衰老IMR90 细胞和肿瘤细胞中可以观察到磷酸化RB 水平降低,并且敲减RB可以显著降低YAP缺失诱导的IMR90 细胞衰老,表明YAP 通过CDK6 调控细胞衰老是RB 依赖的[64]。老年鼠和阿尔茨海默病(Alzheimer disease,AD)模型小鼠的星形胶质细胞以及D-半乳糖或百草枯诱导的衰老星形胶质细胞中YAP 水平都下调并且失活增加。与IMR90成纤维细胞相似,星形胶质细胞衰老的发生也与YAP 调控CDK6 的表达水平相关。除了与正常细胞的衰老有关,YAP调控的CDK6信号通路在肿瘤细胞的衰老中也发挥重要的作用。在胶质瘤的研究中显示,低浓度的β-榄香烯通过抑制YAP-TEAD-CDK6 信号通路,降低了CDK6 的表达,从而诱导胶质瘤细胞发生衰老[76]。同时D-半乳糖也可以通过YAP-TEADCDK6 通路诱导胶质母细胞瘤细胞衰老的发生[77](图3)。
2.3 YAP-自噬 自噬与衰老之间的关系尚存在一定的争议,部分研究表明自噬抑制衰老的发生,但是同时也有研究提示自噬促进衰老的发生。在抑制衰老方面,研究显示衰老细胞的自噬水平明显降低,自噬水平降低后会引起功能缺陷的线粒体在细胞内积累,导致细胞能量代谢障碍及活性氧(reactive oxygen species,ROS)相关的细胞衰老发生[78]。在促进衰老方面,研究表明自噬通过启动细胞衰老来阻止黑色素瘤的发生[79]。同时自噬发生后,在溶酶体的帮助下产生大量的氨基酸,这些氨基酸可用于合成SASP因子来促进衰老的发生[80]。Hippo-YAP信号通路与自噬之间存在广泛的相互作用,并且Hippo信号通路的核心成分MST1、MST2 和YAP-TEAD 都参与了自噬调控,从而在衰老调控中发挥作用。MST1 可以直接磷酸化beclin-1,显着减弱beclin-1-Vps34 和beclin-1-Atg14L的相互作用,在一定程度上抑制了自噬的发生[81]。但是与此同时,MST1/2 也可以通过磷酸化LC3-II 促进自噬的发生[82]。另外,自噬作为YAP-TEAD的下游调控靶标,YAP-TEAD通过影响自噬相关基因的表达影响自噬的水平[83]。Pan 等[6]的研究显示,YAP过表达通过YAP-转录因子阻断自噬或通过激活mTOR 信号通路间接阻断自噬的方式,促进内皮细胞和血管组织的衰老(图3)。
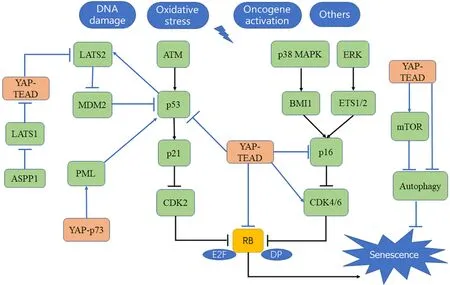
Figure 3. The main mechanism of cellular senescence regulated by Yes-associated protein(YAP). TEAD:transcriptional enhanced associate domain transcription factors;ATM:ataxia-telangiectasia mutated;CDK:cyclin-dependent protein kinase;RB:retinoblastoma protein;MAPK:mitogen-activated protein kinase;ERK:extracellular signal-regulated kinase;BMI1:Bcell-specific Moloney murine leukemia virus insertion site 1;ASPP1:apoptosis-stimulating protein of p53;MDM2:mouse double minute 2;PML:promyelocytic leukemia protein;LATS:large tumor suppressor;mTOR:mammalian target of rapamycin.图3 YAP调控细胞衰老的主要机制
2.4 YAP 调控细胞衰老的其他分子机制 Hippo-YAP 信 号 通 路 可 能 通 过AMPK、mTOR、sirtuin-1(SIRT1)、叉头框蛋白D1(forkhead box protein D1,FOXD1)和转录因子SLUG来调控细胞衰老[84]。作为一种长寿相关激酶,AMPK 的激活可以延长生物模型的寿命[85]。AMPK 与YAP 之间存在广泛的联系,AMPK 可以在多个位点磷酸化YAP 并抑制其转录活性[37],并且在第94 位丝氨酸磷酸化YAP 从而破坏YAP-TEAD 的相互作用[37]。mTOR 可以感受细胞内的营养物质和能量代谢,调节细胞生长及衰老,研究提示消耗mTOR 可以延长中年小鼠的健康寿命[86]。Hippo-YAP信号通路是mTOR 的上游调节因素[87],过表达YAP会增加mTOR 活性,导致血管内皮细胞和血管组织衰老[6]。SIRT1 具有抗衰老和延长小鼠寿命的作用[88]。激活SIRT1 可以引起YAP 发生去乙酰化,减少YAP的激活和核积累,从而中断动脉粥样硬化斑块地形成[89]。同时YAP 也可以激活SIRT1,诱导p53 的去乙酰化,从而抑制p53 相关的G0/G1细胞周期停滞和细胞凋亡,促进A549 细胞存活[90]。YAP与TEAD 转录因子结合后可以激活FOXD1 的表达,从而使衰老的间充质干细胞恢复活力[91]。同时,YAP-TEAD 结合SLUG 转录因子直接抑制促凋亡蛋白BCL-2 修饰因子(BCL-2 modifying factor,BMF)的产生,从而介导非小细胞肺癌衰老样休眠状态[92]。
3 靶向Hippo-YAP治疗衰老相关疾病
3.1 YAP 加重或减轻骨关节炎(osteoarthritis,OA) OA 是一种关节退行性疾病,与细胞衰老存在一定的关联性。膝关节组织中衰老细胞数量的积累与骨关节炎的进展有关[93]。将衰老软骨细胞移植到正常小鼠的骨关节中,该关节也出现了OA 样病理改变和症状[94]。另外,局部清除衰老细胞缓解了创伤后OA 的进展并创造了一个促进软骨再生的环境[15]。目前靶向Hippo-YAP 治疗骨关节炎的研究还处于基础研究阶段。Fu 等[91]的研究提示,YAP 以TEAD 依赖的方式,通过上调FOXD1 来抑制间充质干细胞的衰老。通过关节内注射过表达YAP的慢病毒,可以观察到衰老细胞的数量明显减少,并且减轻了前交叉韧带横断手术构建的OA 模型的病理损伤,减缓创伤后OA 的进展(图4A)。同时,沉默MST1/2或过表达YAP也可以维持小鼠OA 模型的关节软骨完整性,提示YAP 是维持OA 软骨稳态的关键[95]。但是也有部分研究认为YAP 会加重OA 的进展。siRNA 抑制YAP的表达减少了软骨退化和异常软骨下骨形成,改善了OA 的病理表现[96](图4A)。同样的,Zhang等[97]的研究也观察到YAP 在小鼠OA 加重过程中被激活,使得软骨细胞逐渐退化。条件性敲除YAP或使用YAP选择性抑制剂verteporfin 可以保护小鼠OA模型中退化的软骨,并减轻OA 的病理表现。另外,阻断ROR2 受体通过抑制YAP 信号通路也可以减轻OA 模型小鼠的关节疼痛,改善软骨完整性[98](图4A)。总之,Hippo-YAP 信号通路在OA 中的作用尚存在一定的矛盾,还有待进一步的研究予以证实。
3.2 抑制YAP 可减缓动脉粥样硬化的疾病进展细胞衰老(主要是内皮细胞和平滑肌细胞)是导致心血管功能障碍和心血管疾病进展的主要危险因素[99]。内皮细胞和平滑肌细胞的衰老与动脉粥样硬化、动脉瘤、高血压等心血管疾病的进展相关。以动脉粥样硬化为例,衰老的平滑肌细胞会释放促炎细胞因子,导致巨噬细胞等炎症细胞的聚集而加重粥样斑块的炎症,并且释放的基质金属蛋白酶会减少胶原蛋白的含量而促进斑块破裂。同时YAP与动脉粥样硬化的病理生理也具有广泛的联系。相关研究表明,血管内血液的单向层流促进YAP磷酸化,减少YAP 的细胞核转移,进而抑制YAP 下游靶基因CTGF(connective tissue growth factor)和Cyr61(cysteine-rich angiogenic inducer 61)的表达,从而抑制动脉粥样硬化的发生[100](图4B)。靶向Hippo-YAP 治疗衰老相关的心血管疾病是目前研究的热点。在体外培养的细胞和血管组织中使用siRNA 敲低YAP或使用verteporfin 选择性抑制YAP 功能可以缓解自噬阻断,抑制内皮细胞及体外培养的血管组织衰老[6]。同时在分离的粥样硬化动脉组织中使用YAP选择性抑制剂verteporfin 减轻了动脉粥样硬化的程度[101],并且在小鼠体内使用他汀类药物部分抑制YAP激活也可以减轻动脉粥样硬化[102](图4B)。YAP 除了在动脉粥样硬化中具有治疗前景,在心肌再生方面也有一定的研究价值。成人的心肌细胞处于高度分化和增殖停滞的状态,在一定程度上限制了受损心肌的修复。通过重新编程小鼠基因激活YAP,可使增殖停滞的心肌细胞重新进入增殖状态[103]。因此,YAP可能是衰老相关心血管疾病的潜在治疗靶点。
3.3 YAP 可能是治疗AD 的有效靶点 衰老是AD的主要危险因素[104]。衰老的星形胶质细胞随着年龄的增长而积累并导致脑功能障碍和AD 等疾病的发生[105]。Xu 等[66]在体内外敲除星形胶质细胞的YAP,可以通过减少CDK6 的表达水平促进星形胶质细胞衰老;使用MST1/2 抑制剂XMU-MP-1 激活YAP信号部分缓解了星形胶质细胞的衰老并改善了衰老和AD 模型小鼠的认知功能(图4C)。同时AD 小鼠脑组织的基因表达谱分析显示,YAP1是早期AD 相关的最活跃基因,相关细胞实验提示YAP1的表达水平降低可以通过影响整个转录网络来促进早期AD的进展[106]。在小胶质细胞中,使用XMU-MP-1(YAP部分激动剂)或过表达YAP有利于减少β-淀粉样蛋白(amyloid β-protein,Aβ)诱导的促炎细胞因子[107]。在神经元中,Aβ 可以使YAP 转移出细胞核,从而降低细胞核的YAP 水平,引起Hippo 通路依赖的神经元坏死[108]。而使用S1P和YAPdeltaC61(神经元特有的同种型YAP)来增加神经元细胞核YAP水平,可以减轻Hippo依赖的神经元坏死、Aβ聚集和AD小鼠的认知功能障碍[108]。在Aβ1-42诱导的海马神经元凋亡和AD 小鼠模型的研究显示,右美托咪定可以通过miR-129靶向YAP并破坏其与Jagged1蛋白的相互作用,引起海马神经元凋亡减少及AD 模型小鼠认知功能障碍[109]。
3.4 YAP 影响特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)和肝纤维化进展 IPF的患病率随着年龄的增长而增长,并且与肺泡上皮细胞的衰老有关[110]。气管内注射shRNA 的腺病毒敲减YAP1的表达,减少了肺泡上皮细胞的衰老,并且减轻了IPF小鼠的病理改变及临床症状[111](图4C)。与IPF 相反,星状细胞的衰老有利于逆转肝纤维化[112]。四甲基吡嗪(tetramethylpyrazine,TMP)通过抑制YAP 来促进p53 表达,进而促进肝星状细胞衰老,缓解了大鼠的肝纤维化[73](图4C)。在非酒精性脂肪肝炎模型中,选择性阻断巨噬细胞中的YAP 可以减轻啮齿动物和大型动物模型的肝纤维化[113]。而在小鼠缺血再灌注损伤模型中,激活YAP 可以抑制细胞外基质的合成并减少肝星状细胞的激活;抑制YAP 可以增强肝星状细胞的激活,加重肝纤维化[114]。
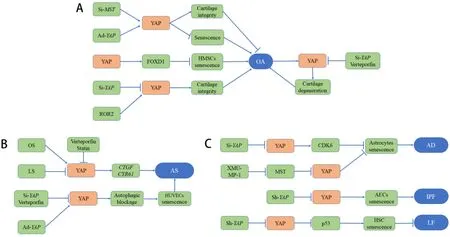
Figure 4. Targeting Hippo-Yes-associated protein(YAP)for the treament of aging-related diseases. A:targeting Hippo-YAP for the treament of osteoarthritis(OA);B:targeting Hippo-YAP for the treament of atherosclerosis(AS);C:targeting Hippo-YAP for the treament of Alzheimer disease(AD),idiopathic pulmonary fibrosis(IPF)and liver fibrosis(LF). MST:mammalian sterile20-like kinase;HMSCs:human mesenchymal stem/stromal cells;HUVECs:human umbilical vein endothelial cells;AECs:alveolar epithelial cells;HSC:hepatic stellate cell;FOXD1:forkhead box D1;ROR2:receptor tyrosine kinase-like orphan receptor 2;OS:oscillatory shear stress;LS:laminar shear stress;CDK:cyclin-dependent protein kinase;CTGF:connective tissue growth factor;CYR61:cysteine-rich angiogenic inducer 61;si-MST:MST siRNA;si-YAP:YAP siRNA;sh-YAP:YAP shRNA;Ad-YAP:YAP adenovirus.图4 靶向Hippo-YAP治疗衰老相关疾病(骨性关节炎、动脉粥样硬化、阿尔茨海默病、特发性肺纤维化和肝纤维化)
4 总结与展望
上游激活因素通过经典Hippo 信号通路或非依赖Hippo 信号通路影响YAP 的磷酸化水平来调控YAP 的活性,从而调控细胞增殖、凋亡和衰老等过程。在调控细胞衰老方面,Hippo-YAP 信号通路通过YAP-ATM-p53-p21、p16INK4A-CDK-RB 和YAP-自噬等多种途径调控细胞衰老的发生,广泛参与OA、心血管疾病、AD 等衰老相关疾病的发生发展,是治疗衰老相关疾病重要的靶点之一。YAP 对细胞衰老的作用因细胞类型的不同而存在差异,可能与YAP 作用于不同的信号通路、YAP 敏感性存在差异、TEAD1~4转录因子存在不同比例等因素有关[18]。深入研究Hippo-YAP 信号通路与衰老之间精确分子调控机制,有利于为衰老相关疾病的治疗靶点提供参考。
