多原子分子简正振动频率的量化计算*
2022-05-26徐又捷郭迎春王兵兵
徐又捷 郭迎春† 王兵兵
1)(华东师范大学物理与电子科学学院,上海 200241)
2)(中国科学院物理研究所光物理实验室,北京凝聚态物理国家研究中心,北京 100190)
3)(中国科学院大学,北京 100049)
针对较大分子振动频率的量化计算,提出了一个节省计算成本的方法.含N 个原子的分子的振动频率的计算通常需要计算3N 维势能超曲面及其二阶导数构成的Hessian 矩阵,然后解其特征方程得到全部简正振动模式的振动频率.N 越大,计算成本越大.本文提出,针对那些由平衡结构和对称性就能完全确定的振动模式,可以逐个计算其振动频率.当仅考虑一个振动模式时,3N 维的Hessian 矩阵的计算转化为一维的势能曲线的计算.基于简谐振子近似推导单一振动模式下分子势能曲线的表达式,接着量化计算势能曲线,将势能曲线拟合到表达式中以获得振动频率.相比计算3N 维势能超曲面及其二阶导数的Hessian 矩阵,仅计算一维势能曲线而节省下来的计算资源可以允许选择更高级别的计算方法和采用更为完备的基组,提高计算的精度.本文首先以计算水分子的B2 振动模式的振动频率为例,说明了这种方法的可行性.接着将这种方法应用到SF6 分子中.多参考组态相互作用(MRCI)方法是计算电子相关能的有效方法,本文采用MRCI/6-311G*基组分别计算了SF6 的A1g,Eg,T2g 和T2u 四个振动模式的振动频率,通过与其他方法的结果以及实验结果相比较,本文计算的四个频率的相对误差最小.
1 引言
量化计算是研究分子结构和光谱的重要工具,振动频率的研究对红外光谱探测具有重要意义.对于N原子分子,量化计算振动频率需要计算3N维的分子势能超曲面及其二阶导数的Hessian 矩阵.多参考组态相互作用(multi-reference configuration interaction,MRCI)等组态相互作用方法因使用多个Slater 行列式近似分子轨道,能够计算更多的相关能[1,2].预期能够得到较精确的振动频率.对于较大分子,由于电子多、基组大,对计算机的内存和速度都有很高的要求,计算成本较大.本文针对较大分子的简正频率计算,提出对3N–6 个简正振动模式分别计算,将3N维的超曲面及其3N维的二阶导数的Hessian 矩阵的计算转化成一维势能曲线的计算.这种计算方法大大降低了计算成本.本文将首先采用较低成本的量化计算方法或对称分析,获得分子振动的简正模式,然后针对单一的简正振动模式,将分子势能表达成单一变量且含有参数振动频率的函数,进而采用MRCI 方法以及较大的基组计算此一维势能曲线,最后拟合势能曲线到势能函数中获得振动频率.
本文在第2 部分将上述方案首先应用到研究比较成熟的水分子的振动频率[3]计算中,来探讨这种方法的可靠性.首先量化计算获得水分子的平衡结构.然后在此平衡结构的基础上,探讨3 个简正振动模式中哪个简正振动模式可以完全确定,哪个简正振动模式除考虑对称性外,还必须考虑力常数.针对可以完全确定的简正振动模式,进行高级别的势能曲线的计算,推导振动频率和势能曲线的关系,进而由势能曲线拟合获得振动频率.最后采用相同的方法,计算基于势能曲面二阶导数构成的Hessian 矩阵,进而得到振动频率,将上面的两个频率结果做比较,确定这种方法的可靠性.
本文在第3 部分将利用此方案计算SF6的振动频率.SF6是被广泛使用的高压电气设备绝缘气体[4−7].SF6分子的振动对SF6分子的高次谐波[8,9]及光电离截面[10,11]等都有直接的影响.人们在实验上[12−16]对SF6的振动频率进行了大量的测量.理论上,采用LDA(local density approximation)/GGA(generalized gradient approximation)泛函密度、MP2 (second-order Moller-Plesset)方法、RCCSD(T)(restricted coupled cluster theory with singles,doubles,and perturbative triple excitations)等方法都对SF6的振动频率进行了计算[17−19].到目前为止,还没有看到采用MRCI 方法计算SF6振动频率的报道.
本文第3 部分安排如下:第1 步,利用分子的对称性以及高级别的计算,获得平衡结构;第2 步,挑选由对称性和平衡结构完全确定的简正振动模式;第3 步,针对第2 步挑选出来的每一个简正振动模式,一方面推导出势能与单一变量之间的函数关系,另一方面,由MRCI/6-311G*对其进行计算,得到势能曲线,然后将曲线拟合到前面推导的公式中,获得每个简正振动模式的振动频率.
2 水分子振动频率的计算
2.1 水分子平衡结构的确定
运用Molpro[20,21]量化计算软件,采用MRCI/def2-TVZPP 对水分子做了几何优化计算.获得水分子的平衡结构为:O—H 键键长l=1.819 a.u.,键角θ=102.755°.
2.2 由分子对称性和平衡结构确定简正振动模式的坐标
由分子的对称性和平衡结构可完全确定分子的一部分简正振动模式的简正坐标.其他的简正振动模式的坐标还需要分子力常数.文献[22]对H2O分子的振动模式的获得做了详细的探讨.这里简述此过程.
H2O 分子属于C2v对称群,C2v对称群有A1,A2,B1和B2四种表示.图1 给出了水分子的3 个核相对其平衡位置的位移构成的九维位移基矢空间,将C2v群的每个操作作用到9 个位移单位基矢上,得到群的一个可约的矩阵表示;进一步将其约化成不可约表示,去掉平动和转动的不可约表示,得到代表振动的不可约表示为:A1,A1和B2.下面将借助投影算符来获得每个振动模式的简正坐标.以B2振动模式为例,将不可约表示B2的特征标投影算符
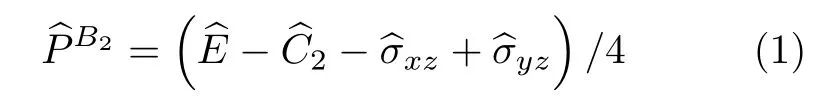
作用在9 个位移基矢单位矢量上(位移坐标系如图1 所示)得到

图1 水分子的位移坐标系示意图,其中1 为氧核,2,3 为两个氢核Fig.1.Schematic diagram of the displacement coordinate system of water molecule.1 represents oxygen nuclei and 2,3 represent the two hydrogen nucleus.

(1)式等号右边的四个对称操作算符依次对应C2v群中的变换操作:恒等变换、绕z轴旋转180°、xz面镜面反射和yz面镜面反射.可见对称性是B2的正则振动矢量可以写作:
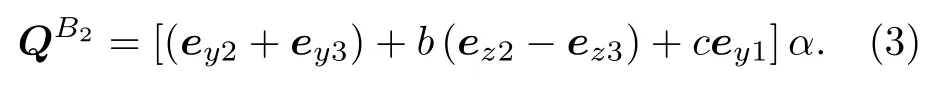
当中b,c是待定系数,α是任一常数.由质心守恒,y方向上有

由角动量守恒条件,x方向上的角动量有
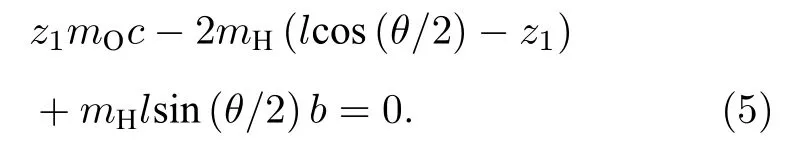
这里mH和mO分别是氢原子和氧原子的质量,z1为 O 原子到质心的距离.于是c=−2mH/mO,b=cos(θ/2),进而可得到对称性是B2的简正振动模式为
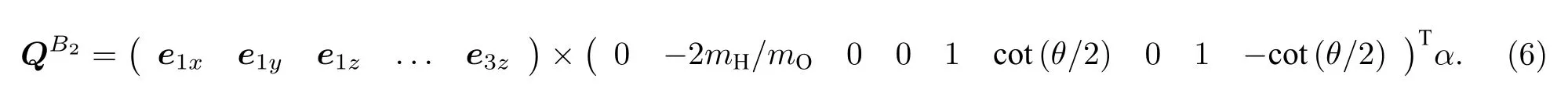
同样可以得另外两个A1简正振动模式为
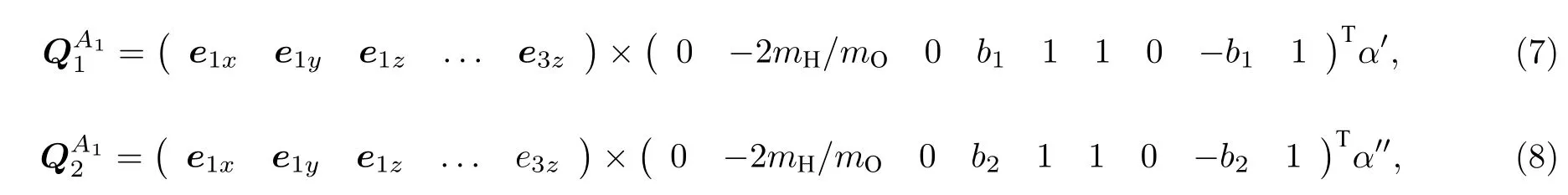
其中α′和α′′为任一常数;b1,b2必须由H2O 分子的分子力常数确定.
由上面求解振动模式的过程可见,分子的振动模式的简正坐标能否由平衡结构和对称性完全确定,与力常数无关,取决于是否存在与其具有不同的频率和相同对称性的其他振动模式:如果存在,则不能完全确定;不存在,则能.我们将计算可以由平衡结构完全确定的振动模式的本征振动频率.对于H2O,将计算B2振动模式的本征频率.
2.3 H2O 分子B2 振动模式频率的计算
本小节中,将针对B2简正振动模式,推导分子势能曲线的表达式.然后结合由Molpro 软件计算出的势能曲线,拟合出此振动模式的振动频率.
2.3.1B2振动模式下的势能表达式
根据波恩-奥本海默(Bonn-Oppenheimer approximation)近似,核在电子的平均势能场中运动.将核的运动近似为平衡位置的简谐振动,则平衡位置附近的势能ε可以表示为
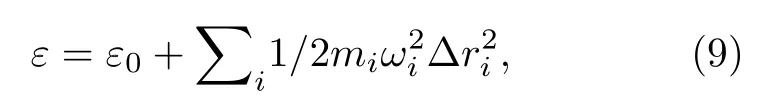
其中ε0代表分子处于平衡位置的能量;i=1,2,3,分别表示氧核和两个氢核;Δri表示第i个核相对其平衡位置的偏离.当只激发一个简正振动模式时,所有核以相同的角速度ω同相振动,振动幅度呈正比关系,比例系数由振动坐标(如(6)式)给出.由此可见,势能的表达式可由一个参量表达.针对水分子的B2振动模式,选取的参量为图1 中2 号H 核的x方向的位移量,记作Δx2,则势能表达为

2.3.2B2振动模式下势能曲线的量化计算
采用MRCI 方法和/def2-TVZPP 基组,针对B2振动模式,即输入核坐标时,核坐标之间的关系满足B2振动模式的简正坐标关系,即满足(6)式.由此得到水分子B2振动模式的势能曲线如图2 所示.图2 中的横坐标是图1 中2 号氢核x方向的相对平衡位置的偏离.

图2 水分子B2 振动模式的势能曲线图Fig.2.Potential curve of of H2O molecule for B2 vibrational mode.
2.3.3 振动频率的拟合计算
将图2 的数据拟合到(8)式中,得到水分子B2振动模式的频率,为3866.18 cm–1.同时利用MRCI方法,采用def2-TVZPP 基组,直接进行频率计算,即计算整个超势能面,计算势能的二阶导数构成的Hessian 矩阵,进而求得H2O 的3 个振动频率,其中包括B2模式的振动频率,为3869.74 cm–1.可见二者之差在几个波数的量级上,是基本一致的.通常理论值和实验值的差别一般在几十个波数以上,所以这个差值是可以忽略不计的.
另外,说明一下拟合过程中偏移量的选择.原则上,偏移量越大,谐振近似越偏离,计算的振动频率误差将越大,我们的目标是计算分子的平衡点处能量的二次导数,即势能曲线底部最小值点的曲率半径.所以将偏移量控制在键长的1%的范围内,计算表明,在此范围内,由于偏移量取值所带来的偏差会在一两个波数的范围内.
关于基组的选择,由于本文拟合的方法需要计算准确的平衡位置(这是获得精确的简正振动模式的振动坐标的要求)和势能曲线,所以要采用尽可能的完备的基组.那么是否可以由基组外推技术来进一步减小误差呢? 采用MRCI/ccPVDZ,MRCI/ccPVTZ 和MRCI/ccPVQZ,针对水分子的B2振动模式,进行了拟合计算,得到相应的振动频率分别为3877 cm–1,3859 cm–1和3882 cm–1,他们并没有随着基组的完备而单调地趋近实验值3943 cm–1.我们没有看到,基组外推能缩小误差.另外,通过考察计算化学数据库[3]所给出的修正因子随基组的完备而变化的情况来探讨这个问题.修正因子(scaled factor)是为了使从头算得到的理论频率更好地和实验值相符合,而将理论值乘以一个在0.8 至1.0 之间的因子.如果基组外推,能够减小频率的偏差,那么,随着基组完备性的增强,修正因子就应该有向1 趋近的趋势,考察由计算化学数据库所提供的修正因子随基组的变化(如QCISD 方法下,基组为3-21G,3-21G*,6-31G,6-31G*,6-31G**,6-31+G**,6-311G*,6-311G**,6-31G(2df,p),6-311+G(3df,2p),对应的修正因子依次为:0.969,0.961,0.964,0.952,0.941,0.945,0.957,0.954,0.947,0.954),没有看到单调的向1的方向趋近的趋势.综上可见,基组外推不能缩小误差.
3 SF6 分子的振动频率计算
接下来把上面的拟合计算振动频率的方法应用于计算SF6分子的振动频率.SF6较H2O 分子复杂得多.它含有70 个电子,具有15 个简正振动模式,所以直接采用MRCI/6-311G* 计算21 阶的Hessian 矩阵而得到振动频率是非常困难的.这里将首先采用MRCI/6-311G*确定分子的平衡结构,然后针对由对称性和平衡结构能完全确定的简正振动模式分别进行频率的拟合计算.
3.1 SF6 的平衡结构
SF6分子是有超高对称性的正六面体分子,如图3 所示.我们唯一需确定的构型参数是S—F 键的平衡键长l0.所以在保持对称不变的前提下,对6 个键长l进行相同的改变,逐点进行MRCI 水平的单点能计算.首先采用HF/6-311G*获得电子组态信息:(core)22(4a1g)2(3t1u)6(2eg)4(5a1g)2(4t1u)6(1t2g)6(3eg)4(5t1u)6(1t2u)6(1t1g)6(6a1g)0(6t1u)0,然后采用完全活性空间自洽场(complete active space self-consistent field)方法确定参考组态.采用的活性空间由1t1g,6a1g和6t1g轨道构成,活性电子数为6 个.最后由MRCI/6-311G*方法获得势能随S—F 键长l的变化曲线如图4 所示,由势能曲线得到SF6的平衡键长为l0=1.558 Å.
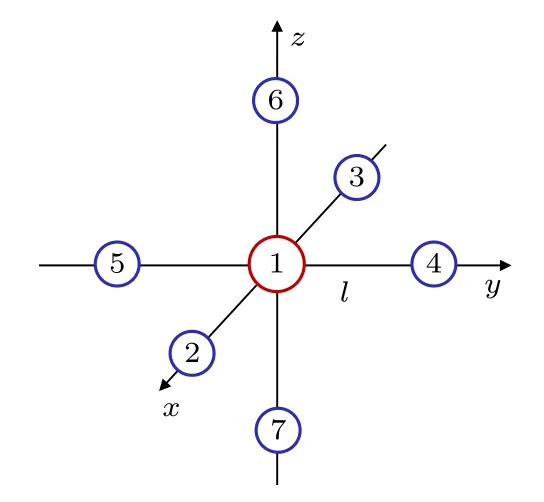
图3 SF6 分子结构示意图,其中 1 为S 核,2—7 为6 个F 核Fig.3.Schematic diagram of SF6 molecular structure. 1 represents Sulfur nuclei and 2–7 represent Fluorine nucleus.
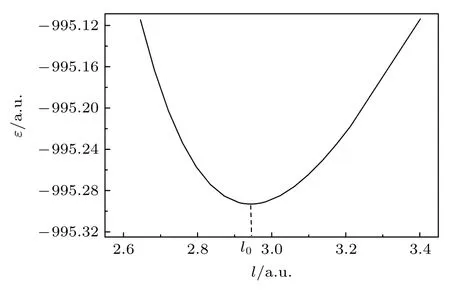
图4 SF6 的势能随S—F 键长l 的变化曲线Fig.4.Potential curve of SF6 as the function of S—F bond length l.
3.2 选择由平衡结构和对称性可以完全确定的简正振动模式
SF6分子所属Oh群.与水分子类似,在SF6分子的7 个原子核相对平衡位置的位移构成的21 维位移空间,约化Oh群的矩阵表示成不可约表示为A1g,Eg,T1u,T1u,T1u,T1g,T2g和T2u,去掉纯转动的T1u和平动的T1g表示,获得SF6分子的15 个简正振动的对称性为:A1g,Eg,T1u,T1u,T2g和T2u.A1g是没有频率简并的,Eg是频率二重简并的.两个T1u,T2g和T2u都是频率三重简并的.对于两个T1u振动模式,它们的频率不同但对称性相同,其简正坐标是依赖于分子常数的.所以本文仅针对A1g,Eg,T2g和T2u四种振动模式进行频率计算.
直接使用投影算符获得SF6分子的简正振动坐标是可行的.但是由于维数多,所以过程繁杂.这里采用量化计算软件最经济的算法HF 方法计算振动频率.虽然这样得到的振动频率的值不是很精确,但是它给出的A1g,Eg,T2g和T2u四种振动模式的简正坐标是准确的,因为它们是由平衡结构和对称性确定的(由于SF6的高度对称性,这四种振动模式的简正坐标与平衡结构也无关),与量化计算的方法和基组的选取无关.A1g,Eg,T2g和T2u四种振动模式的简正坐标列于表1 中,其中下标1 表示S 核,2—7 表示6 个F 核,如图3 所示.
3.3 SF6 的A1g,Eg,T2g 和T2u 振动模式的频率ω1,ω2,ω3 和ω4 的计算
3.3.1 各振动模式势能曲线的表达式
和水分子情况相似,当仅激发A1g振动模式时,由表1 第1 列的振动坐标,SF6的势能为

当仅激发Eg振动模式时,由表1 第2 列的振动坐标,SF6的势能为

由表1 第3 列的振动坐标,SF6的势能为
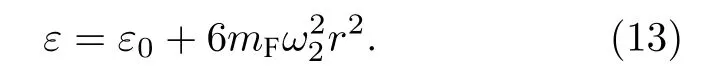
当仅激发T2g振动模式时,由表1 第4 或5 或6 列的振动坐标,SF6的势能为
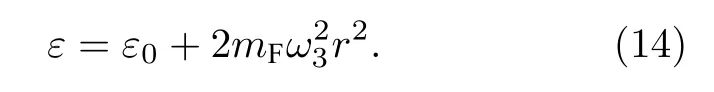
当仅激发T2u振动模式时,由表1 第7 或8 或9 列的振动坐标,SF6的势能为
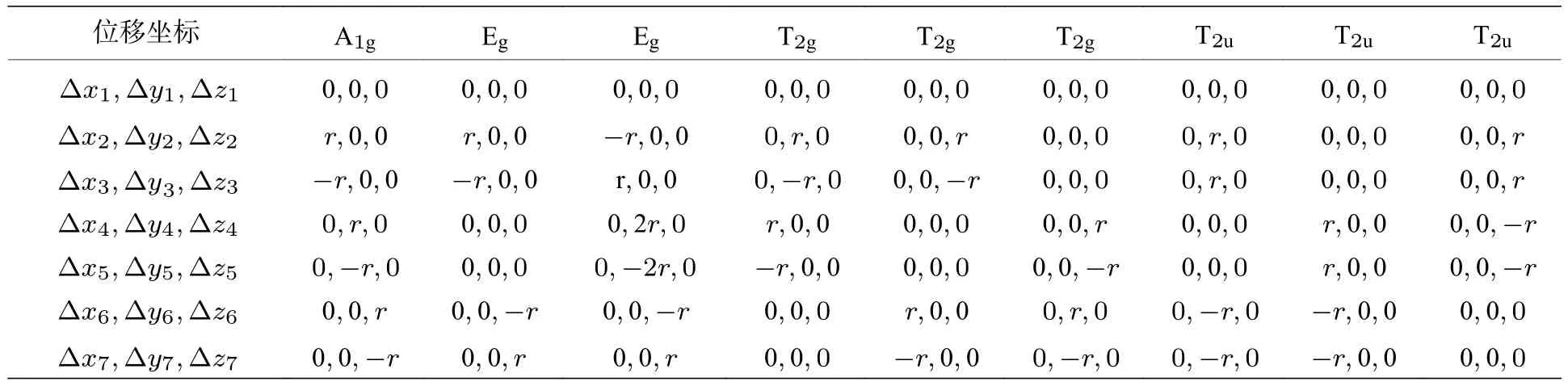
表1 SF6 的A1g,Eg,T2g 和T2u 振动模式Table 1.A1g,Eg,T2g and T2u vibrational modes of SF6 molecule.

3.3.2 各振动模式的势能曲线的从头计算以及振动频率的拟合计算
和2.1 节中获得势能曲线的方法相同,采用MRCI/6-311G*获得各振动模式的势能曲线,如图5所示.将势能曲线数据拟合到(11)式、(12)式、(14)式和(15)式,得到A1g,Eg,T2g和T2u振动模式的频率,具体结果见表2.表2 还给出了利用HF(Hartree-Fock)、CISD (configuration interaction wavefunction including all single and double excitations),MP2,CCSD(T)和B3LYP (Becke,Lee-Yang-Parr)方法由Hessian 矩阵计算的结果,同时还给出了相应方法计算出的平衡S—F 键长l0.我们的目的是比较各个方法的结果,所以采用的基组都是6-311G*.为方便比较,图6 给出了上述不同方法计算的4 种振动频率相对于实验值的相对误差.可见,MRCI/6-311G*方法计算的4 个频率总体来说,相对误差是最小的;平衡键长也最接近实验值.另外,从图6 可见,MRCI 方法获得的4 个频率的连线与其他方法的结果相比,最接近平行于横轴的直线,4 个模式的相对误差起伏最小,MRCI的结果与实验值是最匹配的.

表2 SF6 的A1g,Eg,T2g 和T2u 振动模式的振动频率ω1,ω2,ω3 和ω4Table 2. Vibrational frequencies ω1,ω2,ω3 and ω4 of the vibrational modes A1g,Eg,T2g and T2u of SF6.
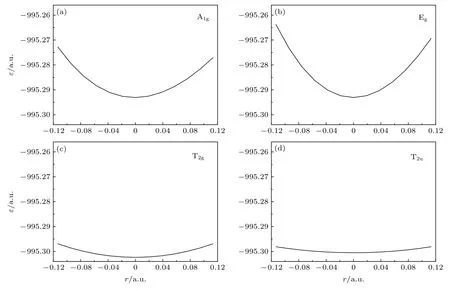
图5 SF6 不同振动模式的势能曲线 (a),(b),(c)和(d)分别对应A1g,Eg,T2g 和T2u 振动模式Fig.5.Potential curves of different vibrational modes of SF6 molecule:(a),(b),(c) and (d) correspond to A1g,Eg,T2g and T2u vibrational modes respectively.
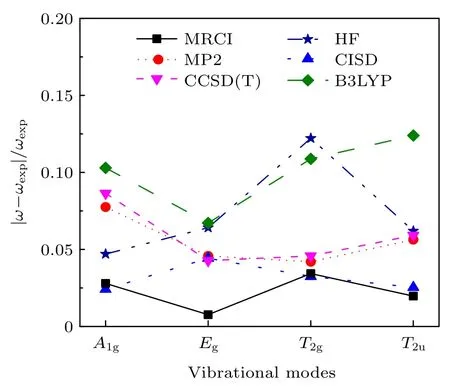
图6 不同方法下使用相同基组6-311G*计算的4 种振动频率相对于实验值的相对误差Fig.6.Relative errors to the experimental values of the four vibrational frequencies obtained by different methods and same basis set 6-311G*.
4 总结
本文给出了一种计算多原子分子振动频率的节约成本的计算方法.即,将计算3N维的势能的二阶导数的Hessian 矩阵分解成计算针对单一振动模式的一维势能曲线,然后推导势能曲线的表达式,通过用表达式拟合势能曲线而得到振动频率.本文的方法适用的振动模式要满足:与其对称性相同而频率不同的振动模式不存在,因为这样的振动模式的简正坐标可以由平衡结构和对称性完全确定,与分子力常数无关.本文计算了水分子的B2振动模式的频率,说明了这种方法的正确性.进而采用这种方法,对SF6的A1g,Eg,T2g,T2u四种振动模式的频率进行了MRCI/6-311G*级别的量化计算.通过和HF,MP2,CISD,CCSD(T)和B3LYP方法以及相同的基组运算的结果以及实验值相比较,MRCI 方法的结果的相对误差是最小的.
