霍山石斛黄酮组分的UPLC 指纹图谱建立及条件优化
2022-05-26刘莉彬祝启张梁汉峰李肖肖宁克壮
刘莉彬,祝启张,梁汉峰,李肖肖,宁克壮,邓 辉
(1. 皖西学院 生物与制药工程学院,安徽 六安 237012;2. 安徽省中药资源保护与持续利用工程实验室,安徽 六安 237012)
大别山区的安徽省霍山县,为石斛属(Dendrobium) 植物分布区域的最北端,其生在多附生于云雾缭绕的悬崖峭壁上和古树上,其中霍山石斛(Dendrobium huoshanense C. Z. Tang et S. J. Cheng)为该地区特有种。随着组培技术和栽培技术的发展,不同产区培育的石斛与霍山石斛在市场上一起流通销售,造成了市场混乱。因此,不同种的石斛或者其他产地的霍山石斛与大别山产的霍山石斛在内在成分上有何区别,需要进行检测确定,以利区分。用超高效液相色谱(Ultra Performance Liquid Chromatography,UPLC) 选取霍山石斛中黄酮组分,建立其最优检测条件,为霍山石斛中黄酮组分的含量和分布差异,提供快速可行的检测方法[1-3]。
1 材料和方法
1.1 仪器设备
HH-4C 型数显恒温水浴锅,金坛市杰瑞尔电器制造有限公司产品;DGX-90738-1 型电热鼓风干燥箱,上海和呈仪器制造有限公司产品;RE-2000A 型旋转蒸发器,中国上海亚荣生化仪器厂产品;FA1204N 型电子天平,沪制MC 精密科学仪器有限公司产品; FS200S-3 新型密封粉碎机,浙江温州瑞安市百信药机器械厂产品;超高效液相色谱仪,美国Agilent 公司产品;二极管阵列检测器,美国Agilent 公司产品;圆底烧瓶;玻璃瓶;回流提取装置。
1.2 材料和试剂
1.2.1 试验材料
霍山石斛,鉴定人:陈乃富教授。
1.2.2 试验试剂
甲醇(色谱纯)、甲醇(分析纯),国药集团化学试剂有限公司提供;乙腈(色谱纯),上海星可生化有限公司提供;乙醇、实验室制双蒸水、娃哈哈矿泉水(色谱用水)。
1.3 试验方法
1.3.1 工艺流程
霍山石斛→烘干→粉碎→过60 目筛→回流提取→蒸发浓缩→微孔滤膜过滤→上样→UPLC 检测→图谱分析。
1.3.2 霍山石斛黄酮组分的提取
取新鲜的霍山石斛鲜条洗净,置于120 ℃烘箱中杀青2 min,再置于70 ℃烘箱中烘干至恒质量。使用粉碎机对烘干的霍山石斛进行粉碎至粉末状,过60 目筛,装于密封袋保存,备用。精密称取1.00 g霍山石斛粉末3 份,分别标为供试品1 号、供试品2 号、供试品3 号,置于250 mL 圆底烧瓶中。供试品1 号中加入70% (V/V) 甲醇100 mL,供试品2 号中加入70%(V/V) 乙醇100 mL,供试品3 号中加入70%(V/V) 丙酮100 mL。70 ℃下水浴回流提取3 h,过滤,取滤液用旋转蒸发仪进行浓缩,收集浸膏待用[4-7]。
1.3.3 霍山石斛黄酮组分的预处理
分别取全部浸膏,用100 mL 甲醇(色谱纯) 溶解,并通过0.45 μm 微孔滤膜过滤,再用0.22 μm微孔滤膜过滤,收集滤液补充至100 mL 备用。
1.3.4 霍山石斛黄酮组分的UPLC 色谱条件优化
进行UPLC 检测时,水(娃哈哈)、甲醇(色谱纯) 和乙腈(色谱纯) 均用0.22 μm 微孔滤膜的超滤仪器进行超滤。检测条件设为UPLC-C18柱,流速0.25 mL/min,检测波长200~400 nm,柱温25 ℃,进样量5 μL,选用乙腈-甲酸水和甲醇-甲酸水系统作为流动相,梯度洗脱,甲醇(10%~90%,0~20 min) -0.3%甲酸水(90%~10%,0~20 min)[8-10]。
2 结果与分析
2.1 提取方法的确立
试验比较了3 种不同的提取方式:冷凝回流提取法、索氏提取法和超声波提取法。因冷凝回流法提取时间短、操作简便,所以最终选用冷凝回流法作为霍山石斛黄酮组分的提取方法。将圆底烧瓶置于70 ℃水浴锅内回流提取3 次,每次1 h,合并提取液。过滤后使用旋转蒸发仪对滤液进行浓缩,浓缩至浸膏状,用少量甲醇(色谱纯) 溶解并通过0.45 μm 微孔滤膜过滤,再用0.22 μm 微孔滤膜过滤,收集滤液,用甲醇(色谱纯) 补充至100 mL 备用。
2.2 检测波长的确立
UPLC 检测过程中,使用DAD 二极管阵列检测器对黄酮组分进行检测,显示在波长200~400 nm 处有出峰,收集出峰液,经化学显色法[11]证明均为黄酮类组分。在波长250 nm 处有最大出峰,但对图谱分析,在波长230 nm 处出峰多,且各峰值总体较高,吸收强度好,区分度明显,无杂峰影响,因此检测波长选择230 nm。
2.3 流动相和洗脱时间的确立
采用甲醇(色谱纯) 和乙腈(色谱纯) 作为流动相洗脱供试品。分别为A(0.3%甲酸水- 甲醇)流动相体系和B(0.3%甲酸水-乙腈) 流动相体系。结果表明,A 流动相体系洗脱分离效果较好。
A 流动相体系洗脱供试品时间见表1,B 流动相体系洗脱供试品时间见表2。
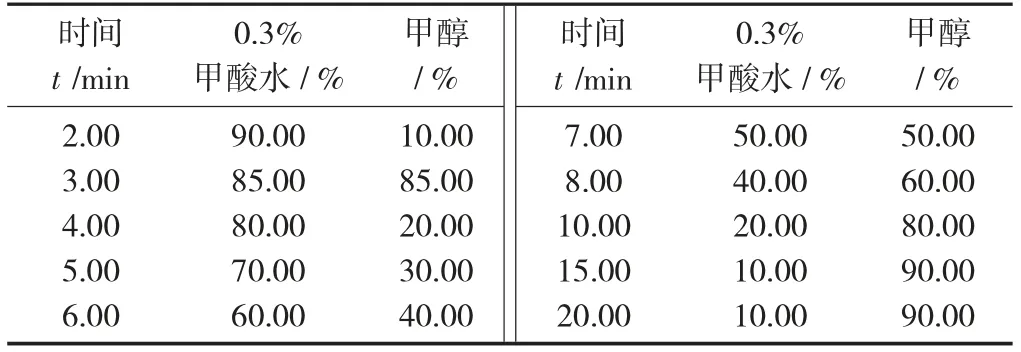
表1 A 流动相体系洗脱供试品时间

表2 B 流动相体系洗脱供试品时间
2.4 指纹图谱的分析
2.4.1 A 流动相体系洗脱供试品1 号
A 流动相体系洗脱供试品1 号的UPLC 指纹图谱见图1,A 流动相体系洗脱供试品1 号的UPLC 指纹图谱3D 图见图2。
由图1 和图2 可知,波长为230 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 000 mAU,在29.985 min 时出现了第2 个组分峰,第2 个组分峰的峰值是2 500 mAU。波长为250 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是500 mAU,在29.985 min 时出现了第2 个组分峰,第3 个组分峰的峰值是1 500 mAU。波长为260 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是550 mAU,在29.985 min时出现了第2 个组分峰,第2 个组分峰的峰值是600 mAU。在200~300 nm 处基线平稳无杂峰。
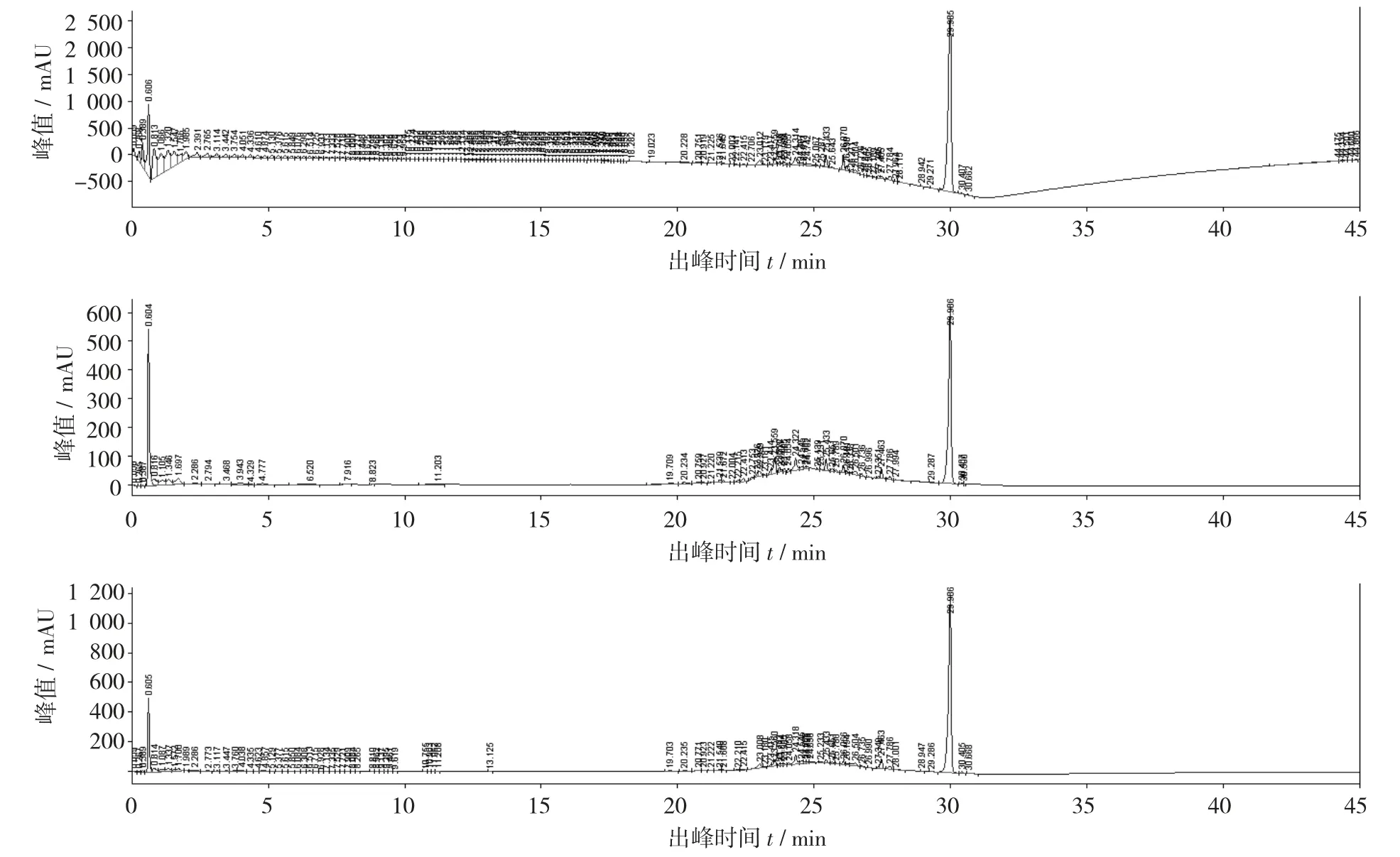
图1 A 流动相体系洗脱供试品1 号的UPLC 指纹图谱

图2 A 流动相体系洗脱供试品1 号的UPLC 指纹图谱3D 图
2.4.2 A 流动相体系洗脱供试品2 号
A 流动相体系洗脱供试品2 号的UPLC 指纹图谱见图3,A 流动相体系洗脱供试品2 号的UPLC 指纹图谱3D 图见图4。
由图3 和图4 可知,波长为230 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 600 mAU,在30.005 min 时出现了第2 个组分峰,第2 个组分峰的峰值是200 mAU。波长为250 nm 时,在0.605 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是600 mAU,在30.005 min 时出现了第2 个组分峰,第2 个组分峰的峰值是200 mAU。波长为260 nm 时,在0.604 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是620 mAU,在30.005 min时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。在200~230 nm 的波长范围内基线漂移,230~270 nm 波长范围内基线平稳,270~400 nm 有杂峰。
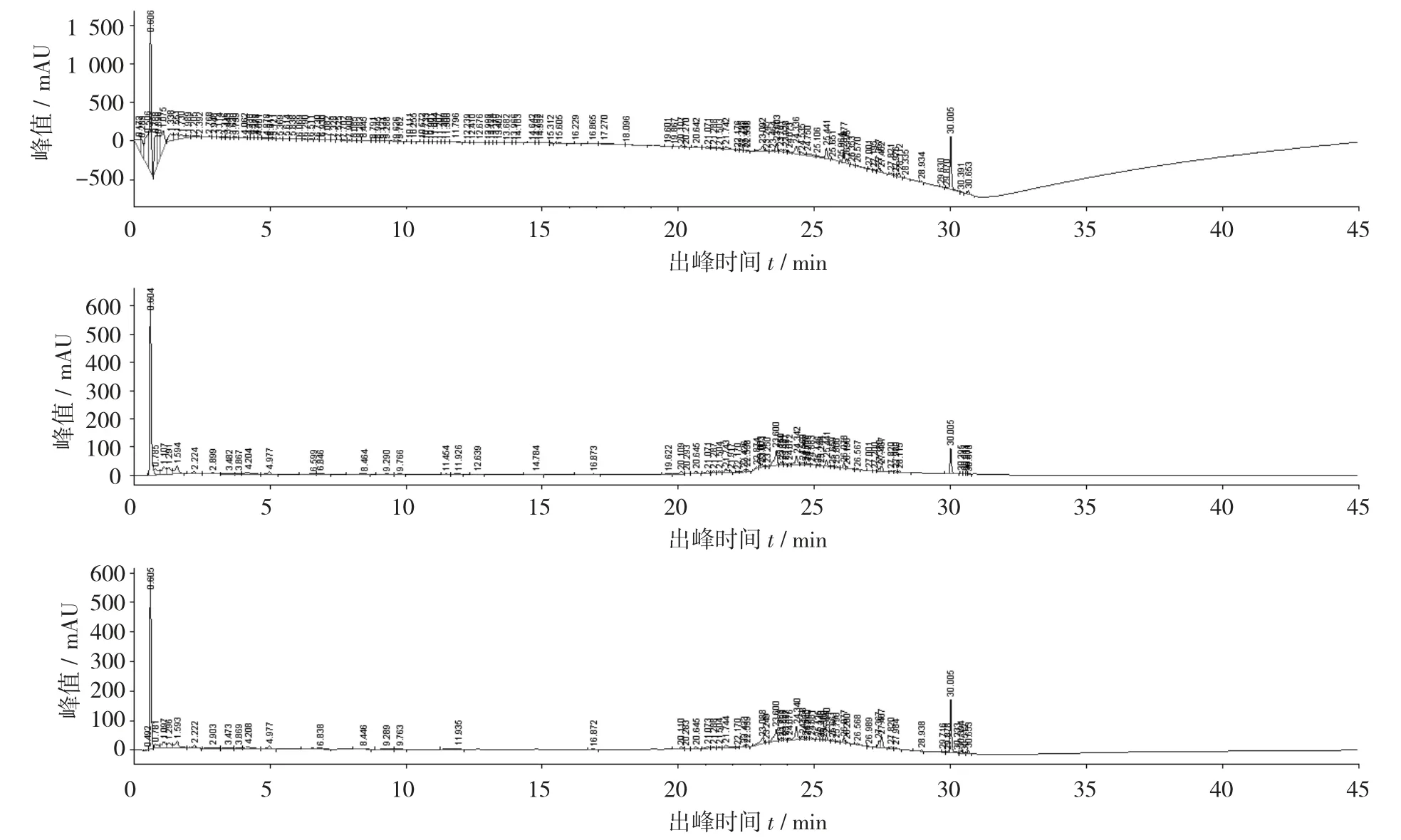
图3 A 流动相体系洗脱供试品2 号的UPLC 指纹图谱

图4 A 流动相体系洗脱供试品2 号的UPLC 指纹图谱3D 图
2.4.3 A 流动相体系洗脱供试品3 号
A 流动相体系洗脱供试品3 号的UPLC 指纹图谱见图5,A 流动相体系洗脱供试品3 号的UPLC 指纹图谱3D 图见图6。
由图5 和图6 可知,波长为230 nm 时,在0.458 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是2 800 mAU,在31.624 min 时出现了第2 个组分峰,第2 个组分峰的峰值是500 mAU。波长为250 nm 时,在0.458 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是2 500 mAU,在31.624 min 时出现了第2 个组分峰,第2 个组分峰的峰值是300 mAU。波长为260 nm 时,在0.459 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是2 400 mAU,在31.624 min 时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。在波长200~220 nm 无第2 个组分峰,波长大于300 nm 时,无第2 个组分峰。

图5 A 流动相体系洗脱供试品3 号的UPLC 指纹图谱

图6 A 流动相体系洗脱供试品3 号的UPLC 指纹图谱3D 图
2.4.4 B 流动相体系洗脱供试品1 号
B 流动相体系洗脱供试品1 号的UPLC 指纹图谱见图7,A 流动相体系洗脱供试品1 号的UPLC 指纹图谱3D 图见图8。
由图7 和图8 可知,波长为230 nm 时,在0.468 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是2 600 mAU,在31.627 min 时出现了第2 个组分峰,第2 个组分峰的峰值是1 200 mAU。波长为250 nm 时,在0.468 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 280 mAU,在31.627 min时出现了第2 个组分峰,第2 个组分峰的峰值是200 mAU。波长为260 nm 时,在0.429 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 300 mAU,在31.627 min 时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。在200~230 nm 波长范围内,基线漂移严重,在230~400 nm 波长范围内,第2 个组分峰峰值低。

图7 B 流动相体系洗脱供试品1 号的UPLC 指纹图谱

图8 A 流动相体系洗脱供试品1 号的UPLC 指纹图谱3D 图
2.4.5 B 流动相体系洗脱供试品2 号
B 流动相体系洗脱供试品2 号的UPLC 指纹图谱见图9,B 流动相体系洗脱供试品2 号的UPLC 指纹图谱3D 图见图10。
由图9 和图10 可知,波长为230 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 600 mAU,在30.005 min 时出现了第1 个组分峰,第2 个组分峰的峰值是200 mAU。波长为250 nm 时,在0.606 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是600 mAU,在30.005 min 时出现了第2 个组分峰,第2 个组分峰的峰值是200 mAU。波长为260 nm 时,在0.604 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是620 mAU,在30.005 min时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。在200~230 nm 波长范围内有杂峰,270~400 nm 范围内有杂峰。

图9 B 流动相体系洗脱供试品2 号的UPLC 指纹图谱

图10 B 流动相体系洗脱供试品2 号的UPLC 指纹图谱3D 图
2.4.6 B 流动相体系洗脱供试品3 号
A 流动相体系洗脱供试品3 号的UPLC 指纹图谱见图11,A 流动相体系洗脱供试品3 号的UPLC 指纹图谱3D 图见图12。
由图11 和图12 可知,波长为230 nm 时,在0.603 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是1 250 mAU,在30.008 min 时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。波长为250 nm 时,在0.602 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是350 mAU,在30.008 min 时出现了第2 个组分峰,第2 个组分峰的峰值是150 mAU。波长为260 nm 时,在0.602 min 时出现了第1 个溶剂峰,第1 个溶剂峰的峰值是350 mAU,在30.608 min 时出现了第2 个组分峰,第2 个组分峰的峰值是100 mAU。在200~400 nm 波长范围内指纹图谱基线漂移严重。
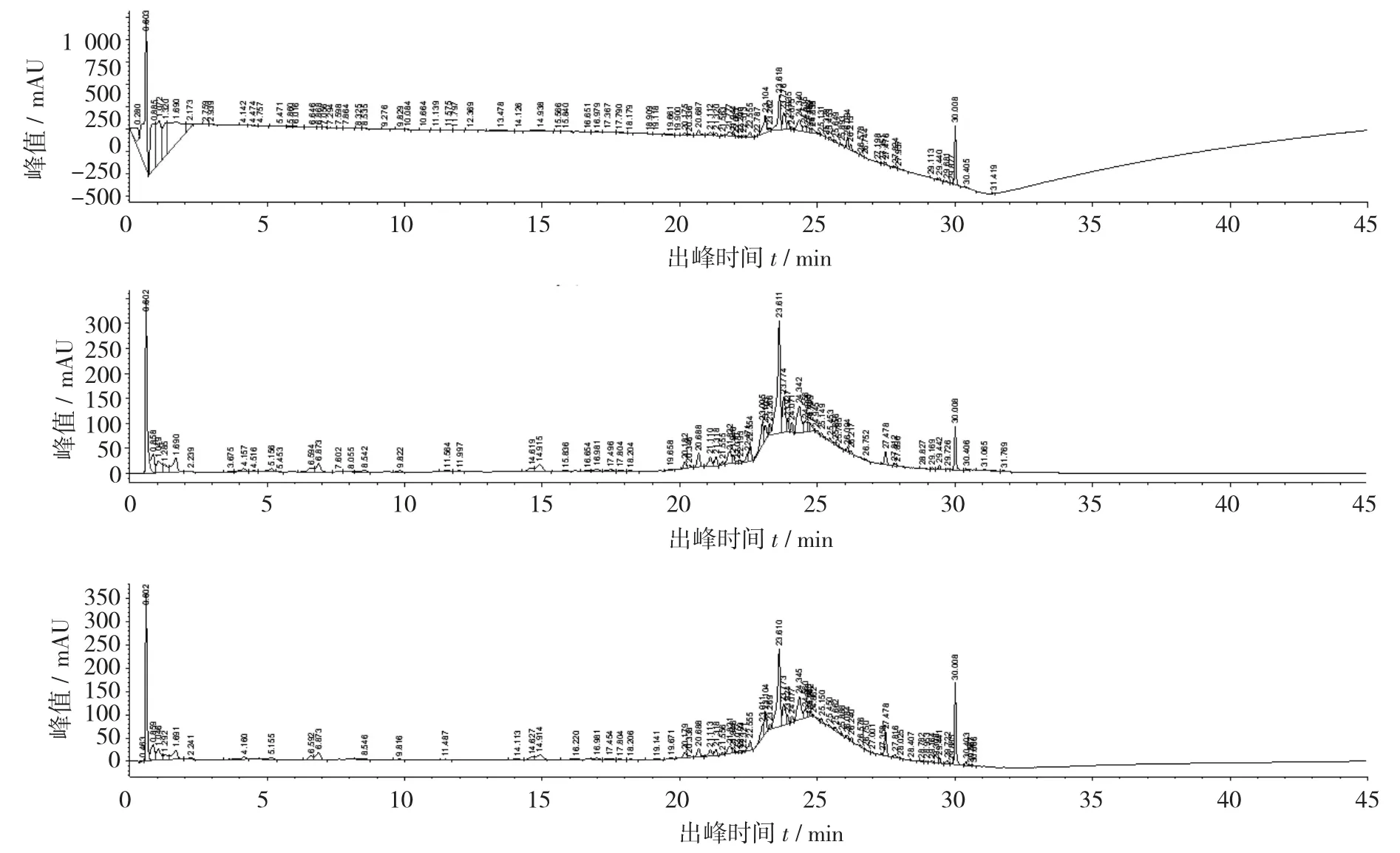
图11 A 流动相体系洗脱供试品3 号的UPLC 指纹图谱

图12 A 流动相体系洗脱供试品3 号的UPLC 指纹图谱3D 图
3 结论
UPLC 分析见表3。

表3 UPLC 分析
试验结果表明,使用冷凝回流法提取出黄酮组分,70%甲醇70 ℃下回流提取3 h 效果最佳,甲醇(色谱纯) 作为流动相洗脱供试品1 号,检测波长在230 nm 处,UPLC 图谱有2 个峰,2 个峰的收集液经化学显色法[11]证明均为黄酮类组分,且2 个峰峰值都较高,图谱的基线平稳。同时,标准品芦丁的出峰时间也靠近30 min。
A 流动相体系洗脱标准品芦丁的UPLC 指纹图谱见图13,流动相体系洗脱标准品芦丁的UPLC 指纹图谱3D 图见图14。

图13 A 流动相体系洗脱标准品芦丁的UPLC 指纹图谱

图14 流动相体系洗脱标准品芦丁的UPLC 指纹图谱3D 图
综上所述,研究建立了专属性强、分离度高、稳定性好的霍山石斛黄酮组分的UPLC 指纹图谱分析鉴别方法,并确定霍山石斛黄酮组分最佳提取方法和洗脱检测方法,霍山石斛黄酮组分UPLC 图谱的建立条件的优化有助于建立霍山石斛相似度评价的黄酮组分共有指纹图谱,并用于霍山石斛的质量鉴定和组效学关系探究,为其质量控制及进一步的药效学研究奠定基础。
