生物信息学方法分析筛选扩张型心肌病发生发展的关键基因和通路
2022-05-21何明俊牟建军
何明俊,白 玲,卢 群,褚 超,任 洁,牟建军
(西安交通大学第一附属医院心内科,陕西西安 710061)
扩张型心肌病(dilated cardiomyopathy, DCM)是引起心力衰竭最常见的疾病之一[1]。DCM 的病因尚不清楚,可能是多因素的,潜在的原因包括感染性、毒性、代谢性、免疫学、神经肌肉性、内分泌性或瓣膜性等。DCM 的特征是心室扩张和收缩功能降低,并导致进行性心力衰竭、左心室收缩功能减弱、恶性心律失常和心源性猝死[2-3]。DCM 可导致实质性的心功能障碍,被认为是心脏移植和/或机械支持的最常见的原因[4]。除获得性DCM 外,20%~48%的DCM 病例是家族性的,可以是常染色体显性或X 连锁。迄今为止,已经在40 多个基因中发现了与DCM 相关的突变[5],主要编码肌节蛋白和细胞骨架蛋白。然而,大多数导致DCM 的变异仍有待确定。本研究拟分析DCM 患者基因测序芯片数据,探讨DCM 患者和正常人心肌组织差异基因和功能富集通路的差异,协助阐明DCM 潜在的分子机制,为诊断和治疗DCM 提供基础。
1 材料与方法
1.1 资料来源
搜索GEO 数据库(http://www.ncbi.nlm.nih.gov/geo),检索关键词为“dilated cardiomyopathy”,“homo sapiens”,“expression profiling by array”。入选标准:①样本取自人心肌组织;②以正常心肌组织样本作为对照;③DCM 组和对照组样本数均大于3 例。基于以上选择,选取数据集GSE42955 和GSE1869 为后续研究序列。GSE42955 的试验平台为GPL6244[HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array[transcript (gene) version],其中包含5 例人正常心肌组织样本,12 例DCM 组织样本。GSE1869 的试验平台为GPL96[HG-U133A]Affymetrix Human Genome U133A Array,其中包含6 例人正常心肌组织样本,21 例DCM 组织样本。在GEO 官网上下载GSE42955 和GSE1869 的基因检测和平台注释数据用于后续研究。
搜索ArrayExpress 数据库,检索关键词为“dilated cardiomyopathy”,设定的筛选条件为organism:“homo sapiens”, experiment type:“all assays by molecule”and“array assay”, By arrays:“all arrays”, 勾选“ArrayExpress data only”,共得到3 个试验结果,选择符合条件的E-TABM-480 数据集为进一步研究数据,应用的测序系统:“Applied Biosystems Human Genome Survey Microarray v2.0”,其中包含4 例正常对照组和5例DCM 患者,样本均取自患者左心室组织,在ArrayExpress 数据库官网上下载E-TABM-480 数据集的基因检测和平台注释数据用于后续研究。
1.2 鉴定差异基因(differentially expressed genes,DEGS)
利用GEO 在线分析工具GEO2R 对数据集GSE42955 和GSE1869 中正常心肌组织和DCM 组心肌组织进行差异基因筛选,筛选条件为P<0.05,设定的FC 切点值为1.3,其中FC≥1.3 为上调,FC<-1.3为下调。利用R 语言(版本4.1.0)ggplot2 函数(版本3.3.5)和ComplexHeatmap 函数(版本2.8.0)分别绘制两组数据集差异基因的火山图和热图。利用R 语言Venn.Diagram 函数(版本1.6.20)计算公共的DEGS并绘制韦恩图。
1.3 蛋白互作网络分析(Protein-Protein Interaction network analysis, PPI)
利用在线数据库STRING(https://string-db.org/)分别对两组数据集筛选所得DEGS 进行PPI 分析。在蛋白互作网络中,一个节点代表一个蛋白质,节点间的连线代表蛋白间的相互作用。使用Cytoscape 软件(版本3.8.2)对PPI 结果进行可视化,并使用软件中的CytoHubba 插件(版本0.1)通过筛选出连接度最高的前10 个基因作为核心基因。使用MCODE 插件(版本2.0.0)鉴定两组数据DEGS 的功能模块,按得分每组数据集取前5 个模块做后续分析。
1.4 模块的功能富集分析
基因本体数据库(GO)分析被广泛用于识别基因、基因产物和序列的特征生物属性,包括生物过程(biological process, BP)、细 胞 成 分(cell components,CC)和分子功能(molecular function, MF)。京都基因与基因组百科全书(KEGG)分析提供了基因组序列和蛋白质相互作用网络信息的全面生物解释。本研究利用R 语言Clusterprofiler 包(版本4.0.2)的enrich-GO 函数和EnrichKEGG 函数对MCODE 插件鉴定得到功能模块进行GO 和KEGG 功能分析,了解模块所执行的生物学功能和相关通路。
1.5 GSEA 分析
GSEA 的富集原理与传统的差异基因富集分析不同,GSEA 分析不需要筛选出差异基因,而利用所有基因及其表达差异进行基因富集分析,富集程度以富集得分(Enrichment Score, ES)表示。本研究分别利用R 语言ClusterProfiler 包(版本4.0.2)的gseGO和gseKEGG 函数对GSE42955 和GSE1869 的正常组和DCM 组的表达数据进行GSEA 分析,并利用Enrichplot 包(版本1.12.2)进行结果可视化。
1.6 差异基因验证
对鉴定得到的核心模块和核心差异基因在ArrayExpress 数据库的E-TABM-480 数据集验证表 达,并 使 用R 语 言ggpubr 包(版 本0.4.0)的compare_means 函数和ggboxplot 函数进行基因差异分析和可视化。
1.7 统计学分析
采用R 语言limma 包进行差异基因表达筛选,设定的FC 切点值为1.3。利用在线数据库STRING 对筛选所得的差异基因进行相互作用分析,参数设置相互作用分数为0.700(high confidence)。使用Cytoscape 软件中的CytoHubba 插件筛选核心基因,方法采取MCC 法,采取得分最高的10 位作为核心基因。使用MCODE 插件进行功能模块鉴定,参数设置为默认参数:Degree cutoff 设置为2,Node Score Cutoff设置为0.2,K-Core 设置为2,Max.Depth 设置为100。采用R 语言的enrichGO 函数和EnrichKEGG 函数进行GO 和KEGG 功能分析,参数设置:qvalue Cutoff设置为0.05。采用R 语言的gseGO 和gseKEGG 函数进行GSEA 分析,参数设置:nPerm=10 000,minGSSize=10,Pvalue Cutoff=0.05。采用R 语言的ggpubr 包的compare_means 函数进行差异基因验证,检验方法采取参数检验(t.test)。所有统计分析采取P<0.05 为差异有统计学意义。
2 结 果
2.1 数据标准化和主成分分析(principal compo⁃nent analysis, PCA)
对GSE42955 和GSE1869 的对照组和DCM 组进行箱线图,样本表达丰度值的分布基本整齐(图1)。PCA 显示,根据PC1 和PC2 值能够较好地区分対照度和DCM 组(图2)。
图1 GSE42955 和GSE1869 数据集基因表达情况Fig. 1 Gene expressions in GSE42955 and GSE1869 datasets
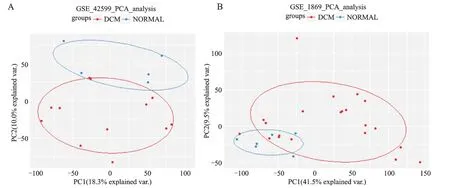
图2 GSE42955 和GSE1869 主成分分析情况Fig.2 PCA of GSE42955 and GSE1869
2.2 鉴别差异基因(DEGS)
按照上述筛选条件,在GSE42955 中筛选出差异基因690 个,其中上调406 个,下调284 个;在GSE1869中筛选出差异基因3 110 个,其中上调1 129 个,下调1 984个。两组的差异基因以火山图和热图的形式分别展现(图3、图4)。使用R 语言的venn.diagram 函数计算共同差异基因,并绘制韦恩图。两组共同表达的差异基因共有110个,其中上调32个,下调78个(图5)。
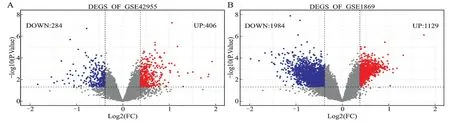
图3 火山图展示两组数据的差异基因Fig.3 Identification of differentially expressed genes

图4 差异表达基因的聚类分析Fig.4 Cluster analysis of differentially expressed genes
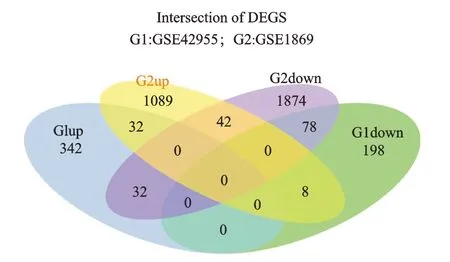
图5 韦恩图展示两个数据集的差异基因Fig.5 Venn diagram shows DEGS between two datasets
2.3 蛋白互作网络分析及核心模块筛选
使用STRING 在线分析工具对纳入GSE42955数据集的690 个差异基因进行功能分析,一共包括326 个点和771 条关系线。GSE1869 数据集由于上述条件筛选得到的差异基因数目过多,无法使用STRING 在线分析工具,按P>0.05 及FC>1.74 或FC<-1.74 再次筛选差异基因,共得到414 个差异基因,一共包括217 个点和452 条关系线。将得到的分析结果分别导入Cytoscape 分析软件,使用Cytohubba 插件分析核心基因,采用MCC 法分析得到GSE42955 数据集核心基因共10 个,分别为SKP2、FBXO32、ASB15、BTBD6、ASB12、FBXL5、ASB8、KLHL41、ASB11、ZBTB16(图6);GSE1869 数据集核心基因共10个,分别为ZBTB16、KLHL2、FBXO21、WSB1、UBA3、UBE2N、CBLB、LTN1、DZIP 3、RNF6(图6)。分别对两组数据集的差异基因使用MCODE插件来分析核心模块,分别得到19 个和14 个核心模块,按得分排序,选择前5 个核心模块进一步分析(表1、表2)。

表1 数据集GSE42955 差异基因得到的核心模块(前5)Tab. 1 Core modules from the dataset GSE42955 differential genes (the first 5)

表2 数据集GSE1869 差异基因得到的核心模块(前5)Tab.2 Core modules from the dataset GSE1869 differential genes (the first 5)

图6 Cytohubba 插件分析两组数据集的核心基因Fig.6 Cytohubba plug-in analysis of core genes in the two datasets
2.4 核心模块功能富集分析
将两组数据集得到的模块基因进行GO 和KEGG 功能分析(图7),两组数据集模块1 的GO 分析显示、功能富集分析显示均为与泛素-蛋白酶体有关的蛋白代谢过程。两组数据集模块1 KEGG 功能富集分析显示,富集分析通路均为泛素介导的蛋白水解(图8)。GSE42955 模块2-5 的GO 和KEGG 分析结果见图9、图10。GSE1869 模块2-5 的GO 和KEGG 分析结果见图11、图12。
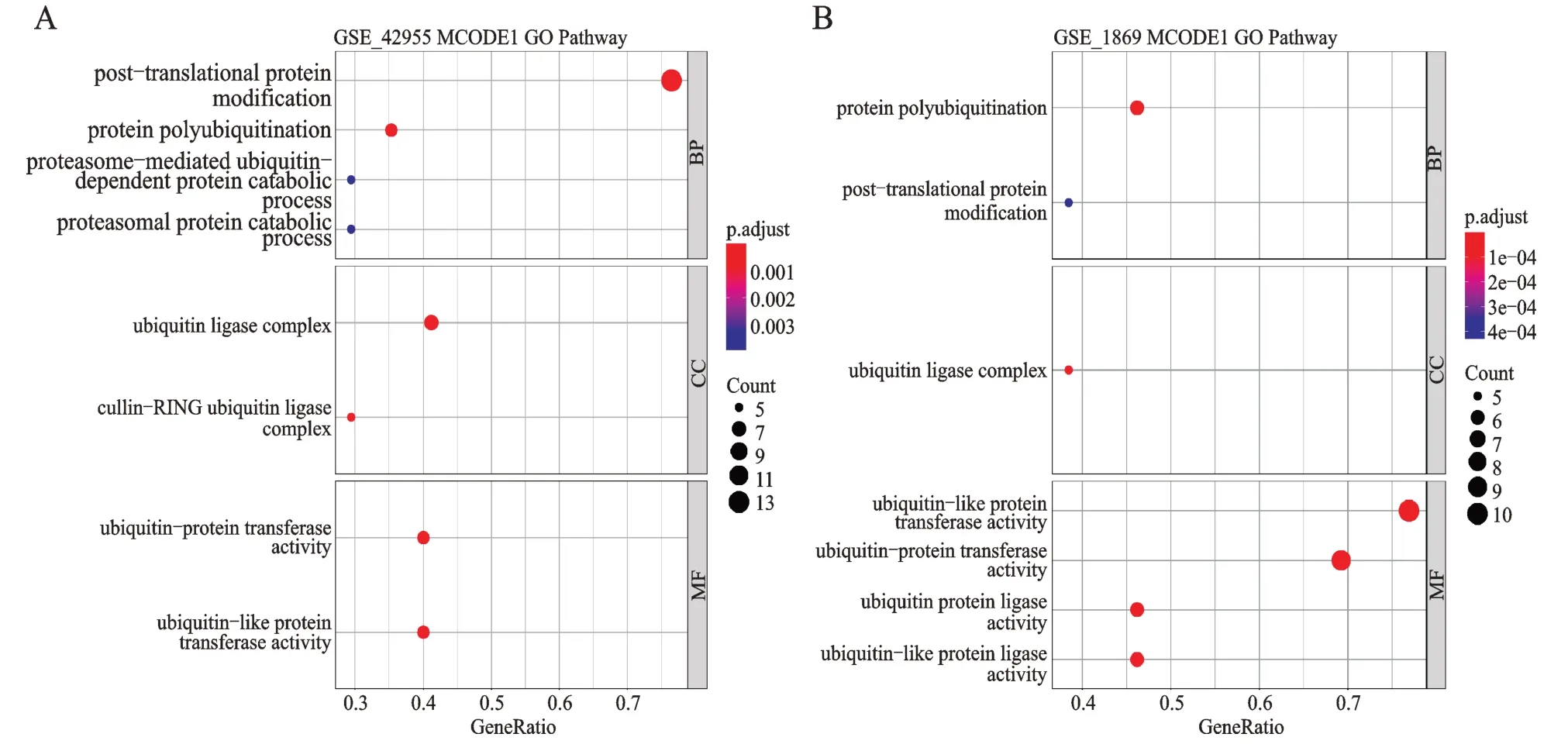
图7 两组基因集模块1 GO 分析结果Fig.7 GO analysis of module1 from the two datasets

图8 两组基因集模块1 KEGG 分析结果Fig.8 KEGG analysis of module 1 from the two datasets

图9 GSE42955 模块2-5 GO 分析结果Fig.9 GO analysis of modules 2-5 from GSE42955
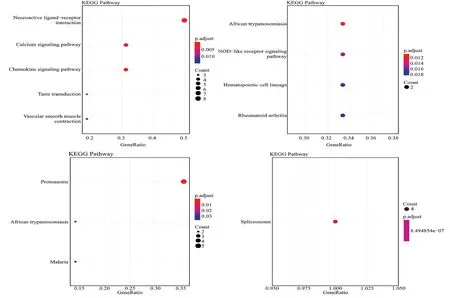
图10 GSE42955 模块2-5 KEGG 分析结果Fig.10 KEGG analysis of modules 2-5 from GSE42955
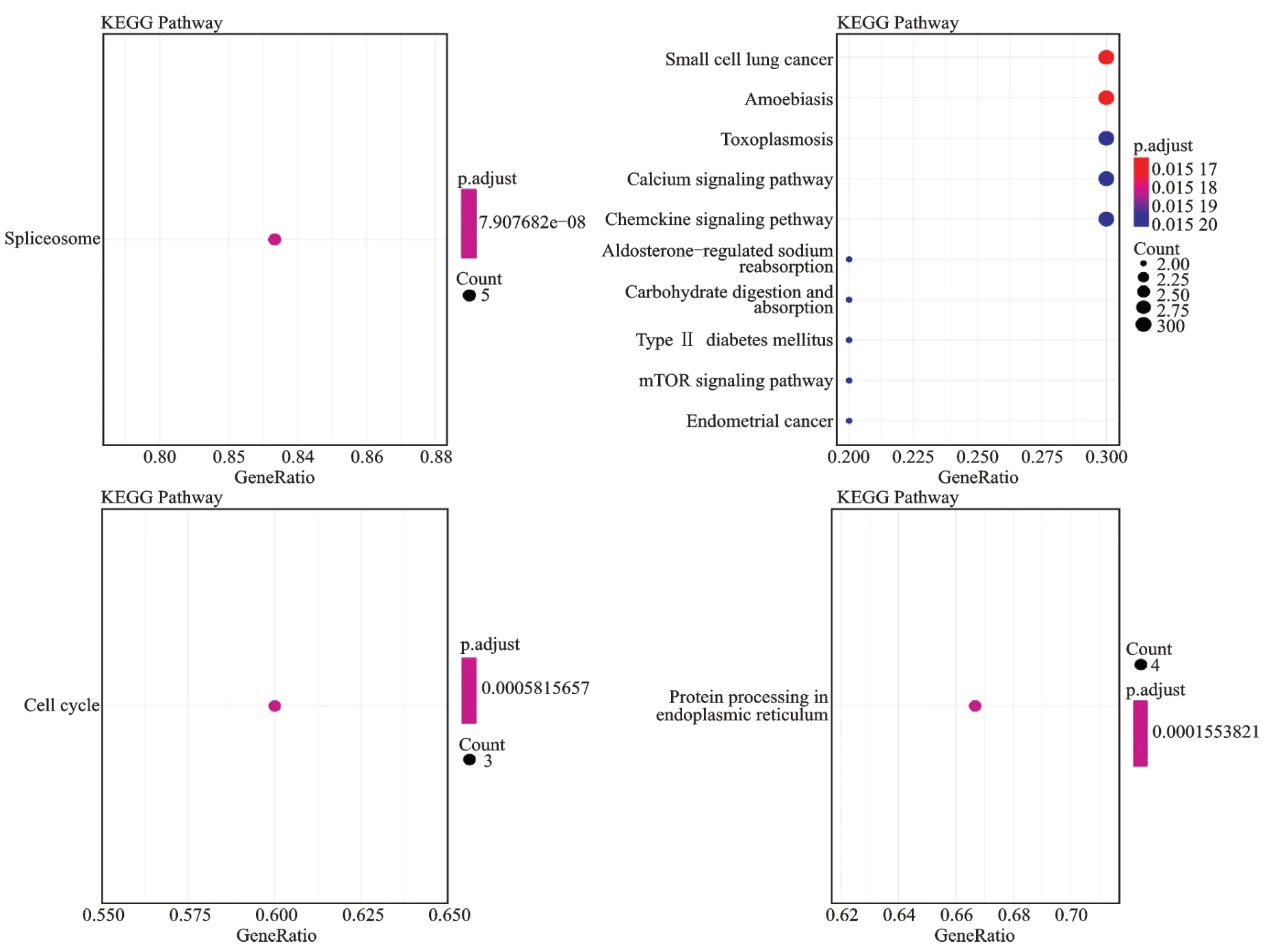
图11 GSE1869 模块2-5 GO 分析结果Fig.11 GO analysis of modules 2-5 from GSE1869

图12 GSE1869 模块2-5 KEGG 分析结果Fig.12 KEGG analysis of modules 2-5 from GSE1869
2.5 GSEA 分析结果
GSEA 结果显示,在对数据集GSE42955 进行GSEA 分析得到的6 条泛素化有关的通路,数据集GSE1869 进行GSEA 分析得到的24 条与泛素化有关的通路,所富集到的通路的NES 值均大于1.5 或小于-1.5,图13 展示的为其中NES 得分和基因富集数较高的6 条通路图。

图13 GSEA 分析结果Fig.13 GSEA analysis results
2.6 差异基因验证
2.6.1 数据标准化和主成分分析 选取对Array-Express 数据库的E-TABM-480 数据集验证表达鉴定到的核心模块和DEGS。对E-TABM-480 数据集表达量绘制箱线图,样本表达丰度值的分布基本整齐,PCA 显示,根据PC1 和PC2 值能够较好区分对照组和DCM 组(图14)。
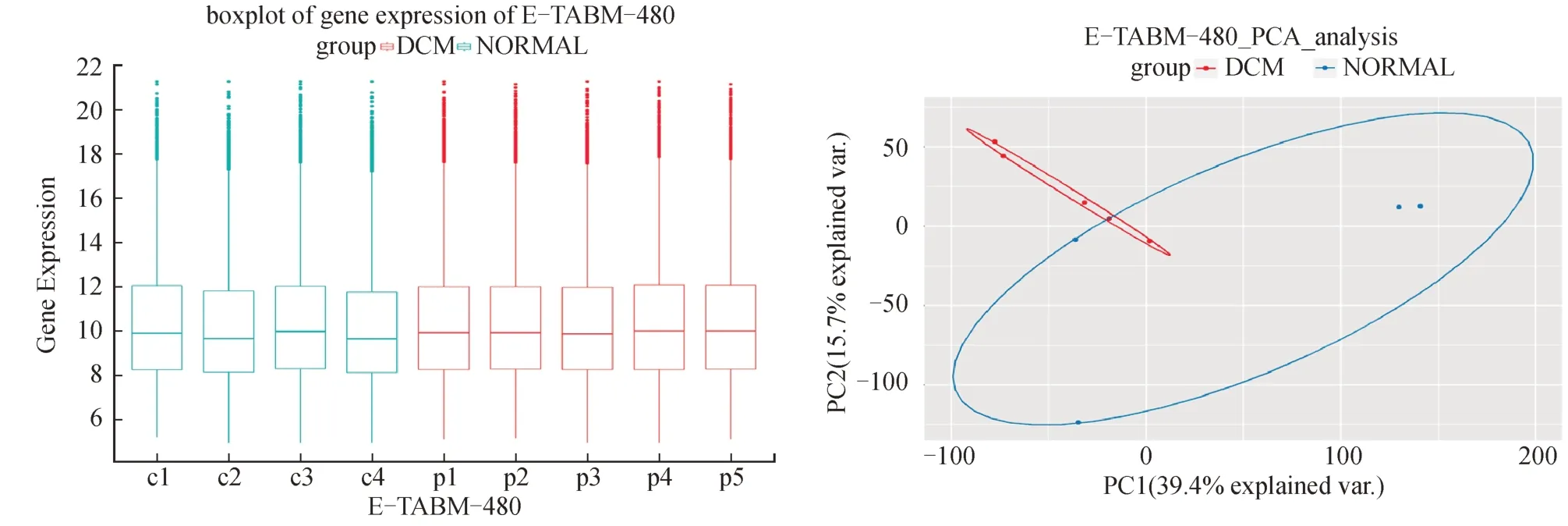
图14 E-TABM-480 数据集样品表达量箱线图和主成分分析Fig.14 Gene expression and PCA of E-TABM-480 dataset
2.6.2 数据集GSE42955 和数据集GSE1869 模块基因表达的验证 对E-TABM-480 数据集进行差异基因分析,按P<0.05,设定的FC 切点值为1.3,DEGS为2 704 个,其中上调为970 个,下调为1 734 个,将3组DEGS绘制韦恩图,可见共表达DEGS共34个(图15)。将筛选出的模块1、模块2 和E-TABM-480 数据集的DEGS 绘制韦恩图(图15),GSE42955 数据集的模块1与E-TABM-480数据集的DEGS 共有6 个共同基因,分 别 为FBXO32、BTBD6、DZIP3、FBXL5、ASB8、COMMD1、GSE1869 数 据 集 的 模 块1 与E-TABM-480 数据集的DEGS 共有5 个共同基因,分别为LTN1、FBXO21、DZIP3、RCHY1、ARIH2,三组共同的差异基因共1 个,为DZIP3,以上DEGS 均与蛋白修饰和泛素功能有关。DEGS 在E-TABM-480 数据集中的表达情况见图16。

图15 韦恩图展示差异基因Fig.15 Venn diagram shows DEGS

***P<0.001,**P<0.01,*P<0.05。
3 讨 论
DCM 是一种常见的心肌病,可导致收缩功能障碍、心力衰竭、恶性心律失常和猝死,并可能发生在疾病的任何阶段[6]。DCM 是进行性心力衰竭的常见原因,也是心脏移植最常见的指征[7]。据估计,特发性DCM 的患病率约为1/2 500,然而加上无症状者,这一比例会更高[8]。在特发性DCM 患者中,20%~50% 的病例是家族性(FDC),FDC 具有遗传异质性[9]。迄今为止,已经在40 多个基因中发现了与DCM 相关的突变[5]。然而,大多数导致DCM 的变异仍有待确定。
利用生物信息学技术筛选疾病基因突变是目前研究的热点方法。本研究从基因数据库GEO 和ArrayExpress 筛 选 数 据,选 取GSE42955、GSE1869和E-TABM-480 三个数据集,基因检测样本均取自DCM 患者或正常心肌组织,利用芯片分析方法检测基因表达数据。通过使用R 语言和生物信息学分析软件分析DCM 和正常心脏组织的差异基因表达和差异生物学富集,以期寻找DCM 发病的关键基因和通路。本研究筛选出的主要差异基因有DZIP3、FBXO32、FBXO21、FBXL5、BTBD6、ASB8、LTN1、RCHY1、ARIH2、COMMD1。DZIP3 基 因 编 码 蛋白,是一种泛素-蛋白质连接酶(E3),以硫酯的形式接受来自E2 泛素结合酶的泛素,然后将泛素直接转移到目标底物上,介导泛素化和随后靶蛋白的蛋白酶体 降 解。FBXO32、FBXO21 和FBXL5 具 有 基 因 同源性,均编码F-box 蛋白家族成员,F-box 蛋白构成泛素蛋白连接酶复合体的四个亚基之一,称为SCF(SKP1-cullin-F-box),FBXO32 在肌肉萎缩期间高度表达,而缺乏这种基因的小鼠被发现对萎缩具有抵抗力,是治疗肌肉萎缩的潜在药物靶点。FBXO32 基因错义突变将影响高度保守的氨基酸序列,会严重损害与SCF 蛋白的结合,AL-YACOUB 等[10]针对扩张型心肌病家系分析显示,FBXO32 基因突变与扩张型心肌病有关。AL-HASSNAN 等[11]的家系分析也显示FBXO32 是隐性DCM 的候选基因,隐性家族性DCM 很少被观察到,通常与心脏外表现有关,如骨骼肌病、肌张力减退、肝性脑病和脂肪酸氧化缺陷受损。FBXO21、FBXL5 也是F-box 蛋白家族的成员之一,FBXL5 相关的疾病包括痉挛性截瘫、常染色体显性遗传和家族性扩张性骨溶解症,重要的同源基因为FBXL13。FBXO21、FBXL5 尚 无 与扩张型心肌病、心肌细胞凋亡及心肌纤维化相关的报道。BTBD6 是一种蛋白编码基因,是一种E3 泛素-蛋白质连接酶复合物的接头蛋白,参与晚期神经元发育和肌肉形成,重要的同源基因为BTBD3。ASB8 是一种蛋白质编码基因,可能是SCF 样ECS(Elongin-Cullin-SOCS-box)蛋白E3 泛素-蛋白连接酶复合物的底物识别成分。LTN1、RCHY1、ARIH2 均为E3泛素-蛋白质连接酶,LTN1 为锌指蛋白,是一种核糖体相关复合物,可介导泛素化和提取未完全合成的新生链以进行蛋白酶体降解。RCHY1 可以介导目标蛋白(包括p53/TP53、P73、HDAC1 和CDKN1B)的E3依赖性泛素化和蛋白酶体降解[12]。ARIH2 与泛素结合酶E2 UBE2L3 一起催化靶蛋白的泛素化。COMMD1 涉及多种生理过程,其功能可能部分与其调节特定细胞蛋白泛素化的能力有关,具有cullin-RING E3 泛素连接酶(CRL)复合物的活性[13],促进NF-κ-B亚基RELA 的泛素化及其随后的蛋白酶体降解而发挥生物学作用[14-16]。上述差异基因的GO 和KEGG 生物学分析显示,生物学过程主要集中在泛素和蛋白酶体相关的生物学过程和通路中。
心脏蛋白质处于不断降解和再合成的动态状态。这个过程高度选择、精确调节,是维持正常细胞功能的关键。其中,溶酶体和蛋白酶体在心脏蛋白质的降解中起重要作用。溶酶体降解大多数胞吞(膜)蛋白,而泛素-蛋白酶体系统(UPS)降解大多数长寿命和短寿命的正常和异常细胞内蛋白质。事实上,哺乳动物细胞中的大部分蛋白质(高达所有细胞内蛋白质的80%~90%)通过UPS 降解[17],因此被认为是细胞内蛋白质降解的主要途径[18]。UPS 由泛素、泛素活化酶(E1)、泛素结合酶(E2)、泛素-蛋白连接酶(E3)、26S 蛋白酶体和泛素解离酶组成。一个功能性的蛋白酶体具有不同的蛋白水解活性、底物和泛素的结合位点[1,19-20]。泛素是一种8.5 ku、高度保守的球状蛋白,附着在其靶蛋白的氨基上[21-22]。泛素分子包含7 个赖氨酸残基,其中标记目标蛋白的特异性赖氨酸残基决定了蛋白质的命运。由泛素激活酶(E1)、泛素偶联酶(E2)和泛素连接酶(E3)组成的级联反应,驱动靶蛋白的泛素化过程。大量的研究已经确定了UPS 在心肌肥厚和心力衰竭中的作用,影响心肌细胞凋亡、细胞质量、肌节质量控制、β-肾上腺素能信号通路、细胞兴奋性和电传导性。它的蛋白质降解活性负责清除90%以上的错误折叠、氧化或其他受损的哺乳动物蛋白,从而防止内在毒性[23]。真核细胞含有一种泛素激活酶E1,但含有多种E2 和E3,底物特异性主要取决于E3连接酶的特性。目前已经鉴定出表达在心脏的E3 泛素连接酶有STUB1(CHIP)、MuRF1、MDM2、Neurl2、FBXO32(Atrogin1、MAFbx)、UBE3A[17]。在疾病的背景下,E3 连接酶表达通过调节信号中间体影响体内缺血再灌注损伤和心脏肥大的疾病严重程度。在缺血再灌注损伤中,CHIP 和MDM2 的活性对细胞凋亡调节和疾病的严重程度产生深远的影响。在心脏肥大中,Atrogin1 和MuRF1 分别通过与钙调神经磷酸酶和PKC 相互作用来减轻心脏肥大。此外,MuRF1、MDM2 与 肌 节 蛋 白(分 别 为cTnI 和Tcap)相互作用,这可能被证明是减轻心脏肥大或调节蛋白质量的机制[24]。
综上所述,本研究利用生物信息学方法鉴定出DCM 在发生发展过程中的差异表达基因和关键通路,鉴定出的差异表达基因和差异富集通路集中在泛素和蛋白酶体相关的生物学过程和通路中,其中FBXO32 已经证实与扩张型心肌病发病相关,但DZIP3、FBXO21、FBXL5、BTBD6、ASB8、LTN1、RCHY1、ARIH2、COMMD1 等基因尚未明确在扩张型心肌病发生发展中的重要作用,值得进一步探索。同时,本研究也表明,生物信息学分析工具在探究心血管疾病发病机制方面的巨大应用价值,相信结合后续分子生物学领域的研究,可以为DCM 的诊断与治疗提供新的思路。
