女性骨关节炎患者外周血单个核细胞基因表达谱的生物信息学分析
2022-05-21袁普卫杜龙龙韩清民
杨 威,袁普卫,杜龙龙,韩清民
(1. 广州中医药大学第三临床医学院,广东广州 510405;2. 陕西中医药大学,陕西咸阳 712046;3. 广州中医药大学第三附属医院,广东广州 510405)
骨关节炎(osteoarthritis, OA)是一种以软骨不可逆性退变为主的最常见关节病,使受累关节负荷疼痛和功能受限,好发于中老年女性,严重影响患者生活。OA 主要病理特征为关节软骨降解、滑膜炎症及其周围组织重塑[1],与遗传、衰老及慢性炎症等多因素有关,但其病因病机不明。随着人口老龄化及肥胖加剧,患病率持续增长已成全球共识。目前,临床没有监测OA 发生或进展的特异靶点,不仅缺乏对OA 的早期诊断,也无减缓疾病进展的方法。因此,探索对OA 敏感且实用的分子标志物十分迫切。而血液作为临床信息的重要来源,提供包括代谢物、蛋白质和核酸等信息,随着分子生物学的发展,与OA 相关的血液生物学指标也逐渐增多,尤其是关注外周血单个核细胞(peripheral blood mononuclear cells, PBMCs)的生物标志物。已有国内外研究鉴别出OA 患者PBMCs基因表达谱中可能用于诊治的差异基因[1-4],但都是对所有OA 患者PBMCs 基因分析,没有区分性别,也没有采用基因集合富集分析(Gene Set enrichment analysis, GSEA),而且所有分析结果都不一致。
本研究分析数据集中所有女性外周血样本,基于基因集富集分析(gene set enrichment analysis,GSEA)寻找与OA相关的目的基因,去重后进行经典的基因本论(gene ontology,GO)富集、京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)通路富集和蛋白互作可视化网络(protein-protein interaction network,PPI network)分析,运用MCODE、cytohubba 和centiscape 插件筛选核心基因,为寻找OA 诊治的适宜分子靶点提供理论基础。
1 资料与方法
1.1 数据来源
从GEO(https://www.ncbi.nlm.nih.gov/geo)数据库查找OA 患者PBMCs 基因表达芯片GSE48556[2],包括106 例OA 患者血液样本和33 例正常志愿者(NC)样本,由于仅1 例男性OA 患者,样本量过少,无法进行性别对比,故予剔除。本研究针对数据集中所有女性患者,纳入情况见表1,所处平台为GPL6947 Illumina HumanHT-12 V3.0 expression beadchip,类型为表达谱芯片。本研究共有48 802 条数据,25 159个基因。
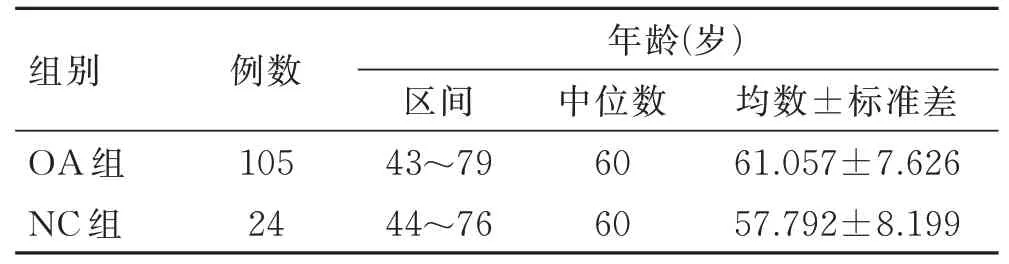
表1 纳入样本情况Tab.1 Characteristics of the included samples
1.2 数据质量与差异分析
用GEO2R(http://ncbi.nlm.nih.gov/geo/geo2r)对数据集进行分组及分析,使用R 语言的limma 包对样本进行一致性检验和主成分分析(principal component analysis, PCA),用以检测样本的均一性、可重复性和组间差异性。
1.3 GSEA 分析
将原始数据经R 语言计算出相应的P.adjust、qvalue、Pvalue 和log2基因表达差异倍数(fold-change,FC)等,用clusterProfiler 包进行GSEA 分析,于分子特征数据库官网(http://www.gsea-msigdb.org/gsea/msigdb/index.jsp)分别选择基因集C2.CP.KEGG.v7.4、C5.GO.BP/CC/MF.v7.4做相应分析。用GSEA分析找到富集的生物学过程或通路,将对应的所有leading-edge 基因累加后去重。
1.4 目的基因筛选
将得到的leading-edge 基因子集用excel 2010 在经R 语言预处理过的原始数据中找到相对应的P.adjust、qvalue、Pvalue 和log2FC 值,筛 选 条 件 是P.adjust<0.05&log2FC|>0.2[5],共得到目的基因292 个。
1.5 GO 和KEGG 富 集 分 析
使用DAVID 数据库(https://david.ncifcrf.gov/tools.jsp)对筛选出的292 个基因进行GO 和KEGG通路富集。GO 分析包括生物学过程(biological process, BP)、细胞成分(cellular component, CC)和分子功能(molecular function, MF),用于预测蛋白质的功能。KEGG 通路分析出分子间相互作用、反应和构建关系网络。P<0.05 认为差异具有统计学意义。
1.6 PPI 网络构建及模块分析
将目的基因输入STRING(https://string-db.org/)在线平台,设置combined score>0.4,然后将计算结果导入Cytoscape-v3.8.2 中,构建PPI 网络。通过MCODE 插件分析出PPI 网络中连接最为紧密的基因集簇,使用cytohubba 插件采用MCC[3]和Degree算法筛选出连结度最高的前10 个基因,再联合centiscape 插件中Degree、Betweenness、Closeness 三个参数以及客观的P值、P.adjust 和log2FC 值综合分析筛选核心基因。
2 结 果
2.1 数据质量与差异分析
将GSE48556 数据集所有女性分为OA 组和NC组,分别有105、24 例,标准化后各样本中位数基本在一条水平线上(图1)。二维及三维PCA 分析,分组间差异不够明显,说明PCA 对组间差异解释有限(图2)。ggplot2 包制作全基因组火山图,FC 值设定为1.2,P<0.05 有统计学意义,显著上调有326 个基因,显著下调有385 个基因,ComplexHeatmap 包制作热图,显示表达谱中高、低表达各top20 基因(图3)。
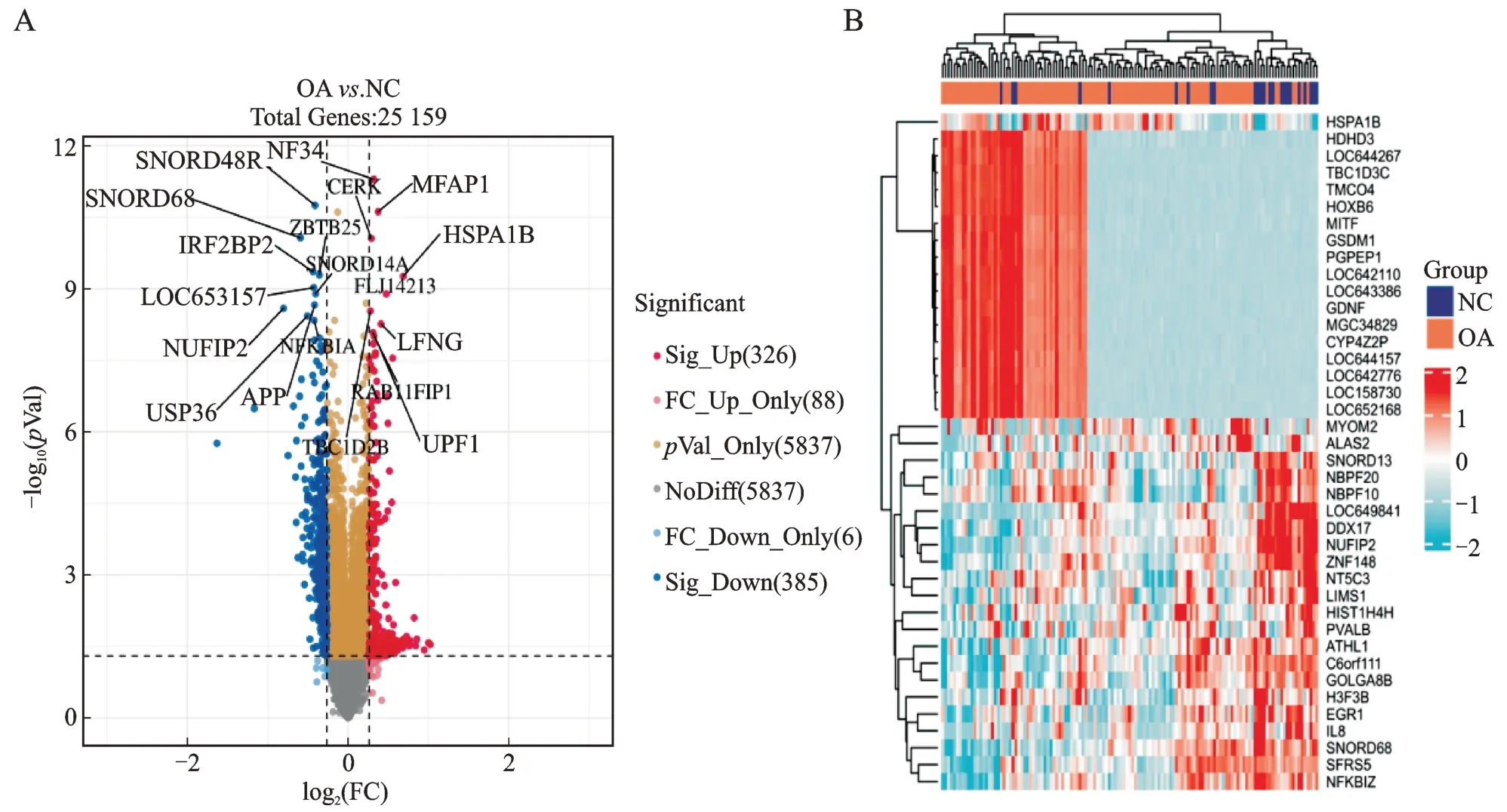
图3 GSE48556 数据集差异表达的火山图和热图Fig.3 The differentially expressed volcano map and heat map of the GSE48556 data set

A、B 分别为归一化前后,绿色为NC 组代表正常对照,蓝色为OA 组。
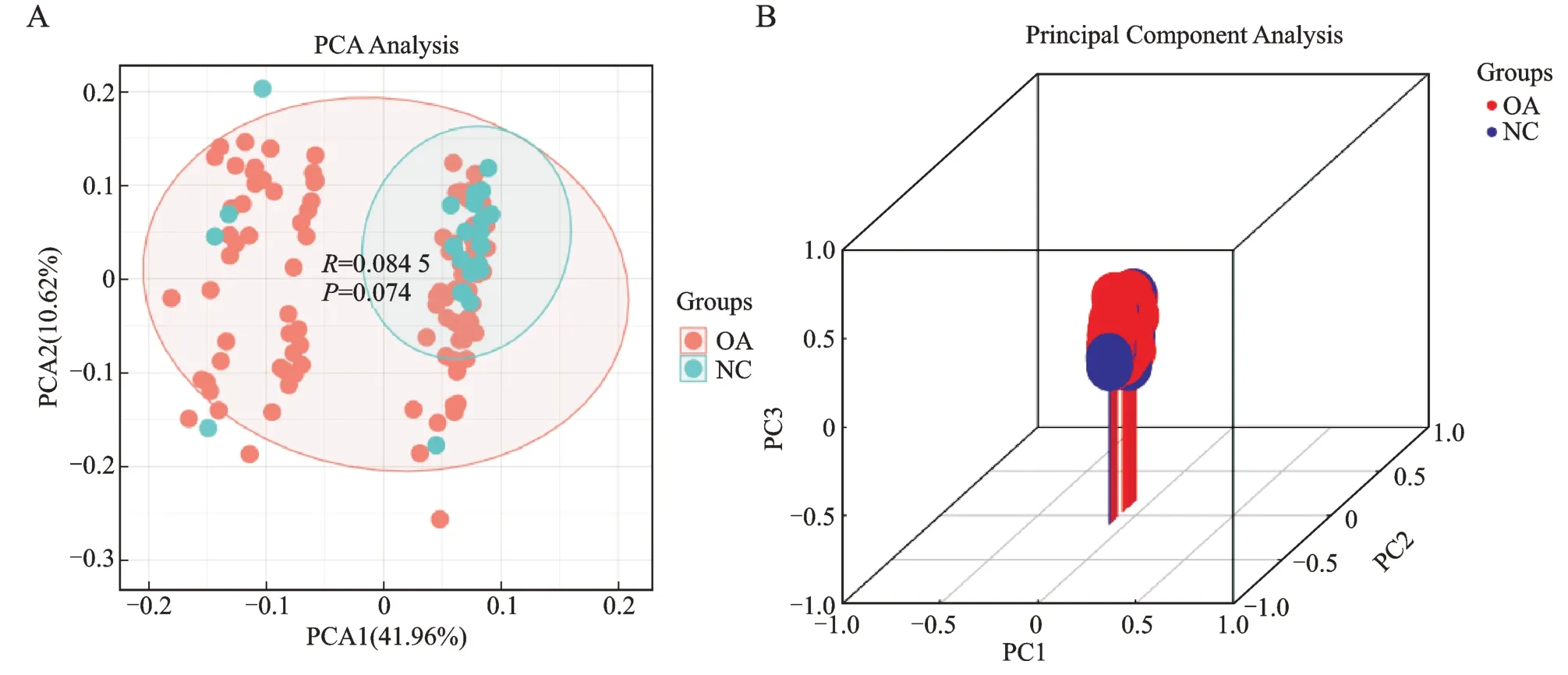
A、B 图分别为二维或三维,PC1、PC2、PC3 分别表示主成分1、2、3,NC 为正常对照组。
2.2 GSEA 分析
GSEA 分析找到富集的生物学过程或通路,筛选条件为:BP 和CC 以P.adjust<0.05 &qvalue<0.25,MF 和KEGG 以P<0.05 &qvalue<0.25,分别筛到80、37、22、16 条,将对应的所有leading-edge 基因累加后去重,一共得到3 120 个基因。富集到的BP 多与固有免疫和适应性免疫调节等有关;CC 多为细胞核仁、核膜、囊泡和内质网等;MF 多与酶、转录因子或组蛋白等的结合有关;KEGG 主要与移植物抗宿主病、抗原呈递和自然杀伤细胞介导的细胞毒性等有关,以下分别展示与OA 相关的功能或通路(图4)。
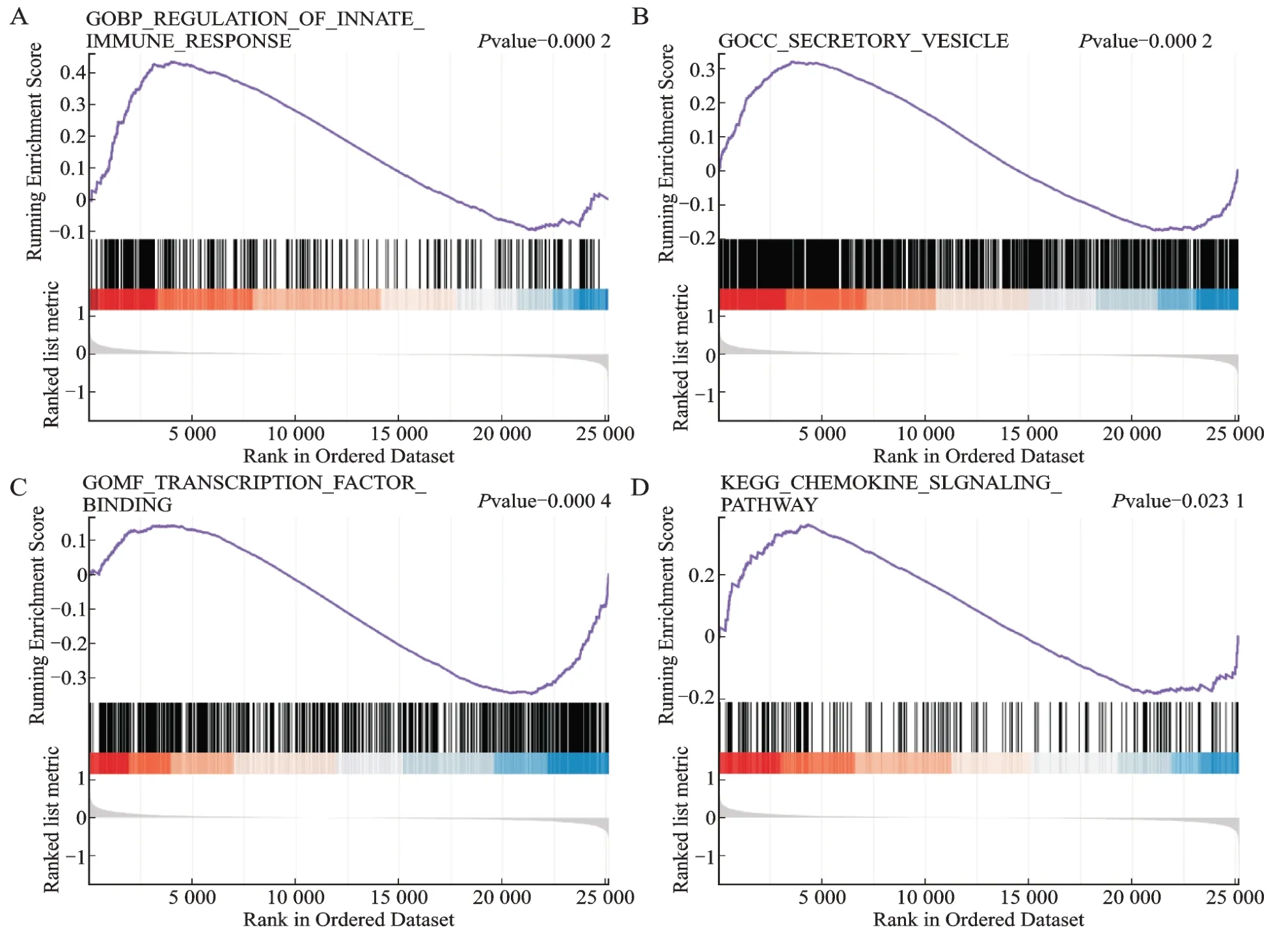
图4 基因芯片GSE48556 数据集GSEA 富集分析Fig.4 GSEA enrichment analysis of gene chip GSE48556 data set
2.3 目的基因
经过筛选得到的leading-edge 基因子集,有3 120个基因,其中上调基因1 195 个,下调基因1 925 个;考虑到功能基因的差异和显著性,满足P.adjust<0.05 & |log2FC|>0.2 的有292 个目的基因,其中上调81 个,下调211 个,分别做热图对比展示(图5)。
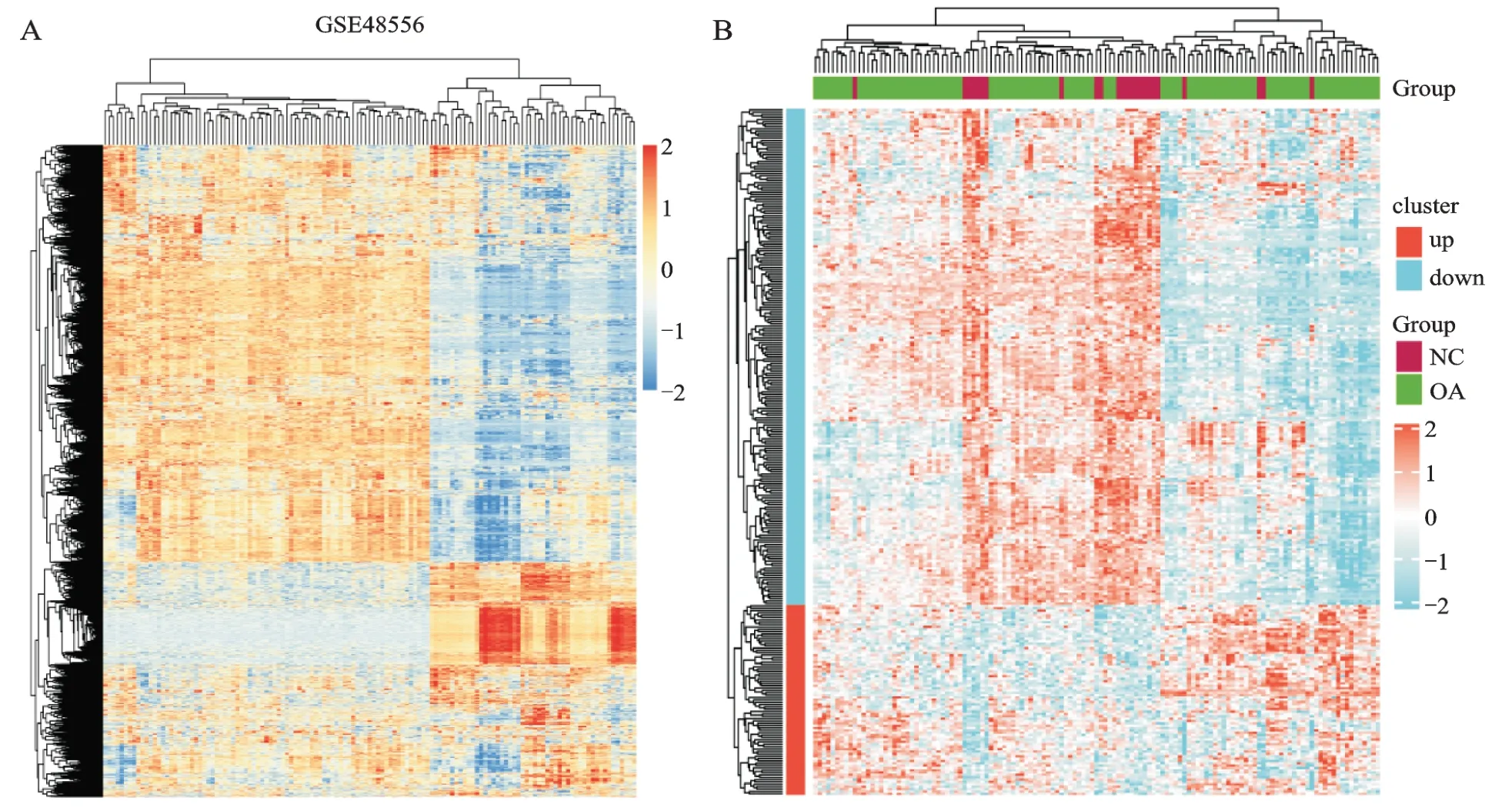
图5 leading-edge 基因子集和目的基因热图Fig.5 The heat maps of leading edge gene subset and target genes
2.4 GO 功能和KEGG 通路富集分析
对目的基因进行GO 和KEGG 富集分析,共富集到289 条,其中在满足P.adjust<0.05 &qvalue<0.2条件下,BP 共有124 条,CC 共有12 条,MF 共有8 条,KEGG 共有11 条。BP 主要富集在NIK/NF-κB 信号的调节、单核细胞增殖、凋亡信号通路的调控、TNF介导的信号通路和Wnt 信号通路的调控等;CC 富集在细胞核、核膜和胞质连接等;MF 富集在蛋白质结合、分子接头、细胞趋化因子受体活性或调节等;KEGG 富集在自然杀伤细胞介导的细胞毒性、TNF信号通路、MAPK 信号通路、细胞凋亡、T 细胞受体信号通路、FoxO 信号通路、NF-κB 信号通路和趋化因子信号通路等。将P.adjust 的值从小到大排序,结合与OA 的相关性展示BP 和KEGG,分别有12 条和10 条,CC 和MF 全部展示,见表2、图6。
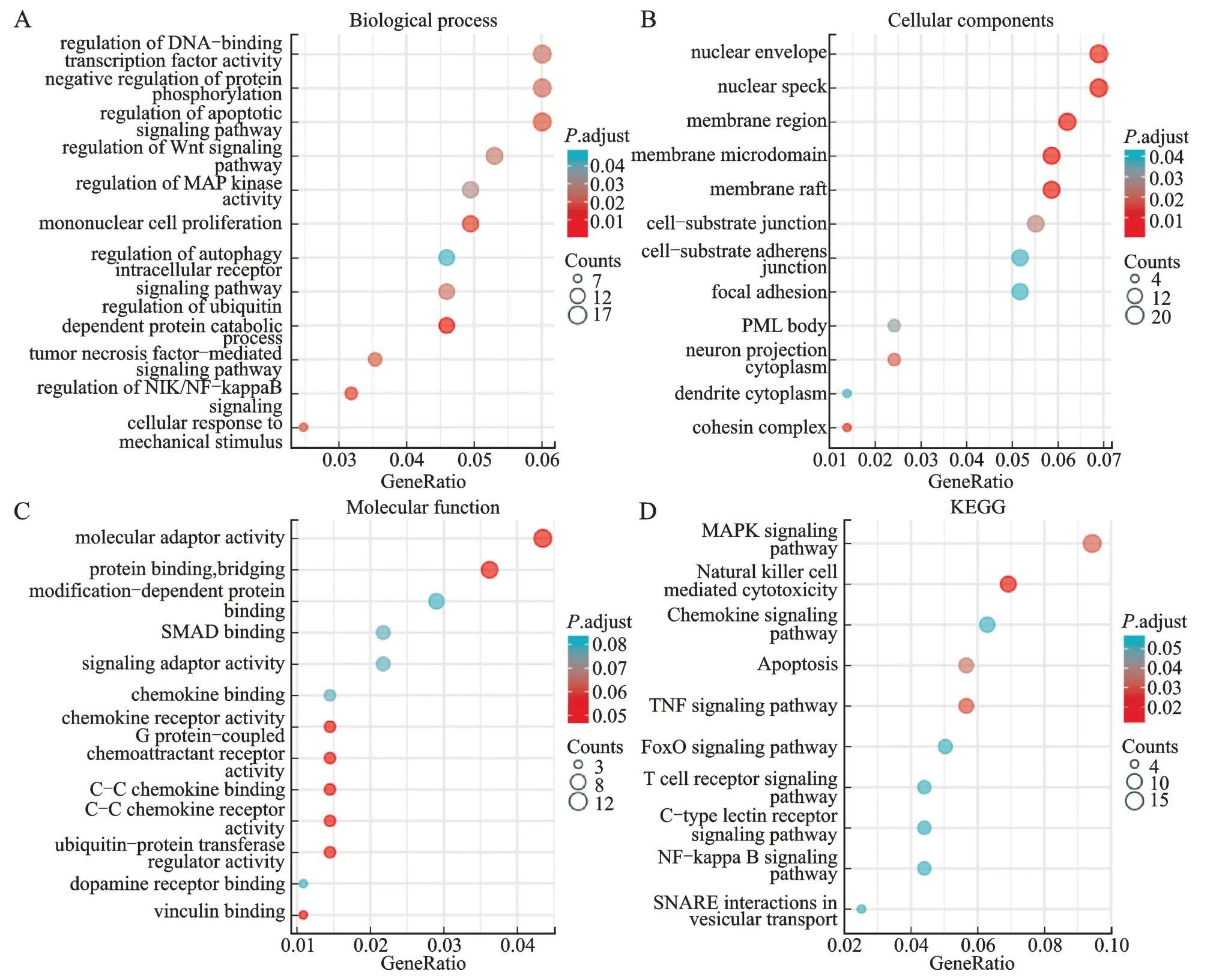
图6 GO 和KEGG 富集分析气泡图Fig.6 Bubble charts of GO and KEGG enrichment analyses
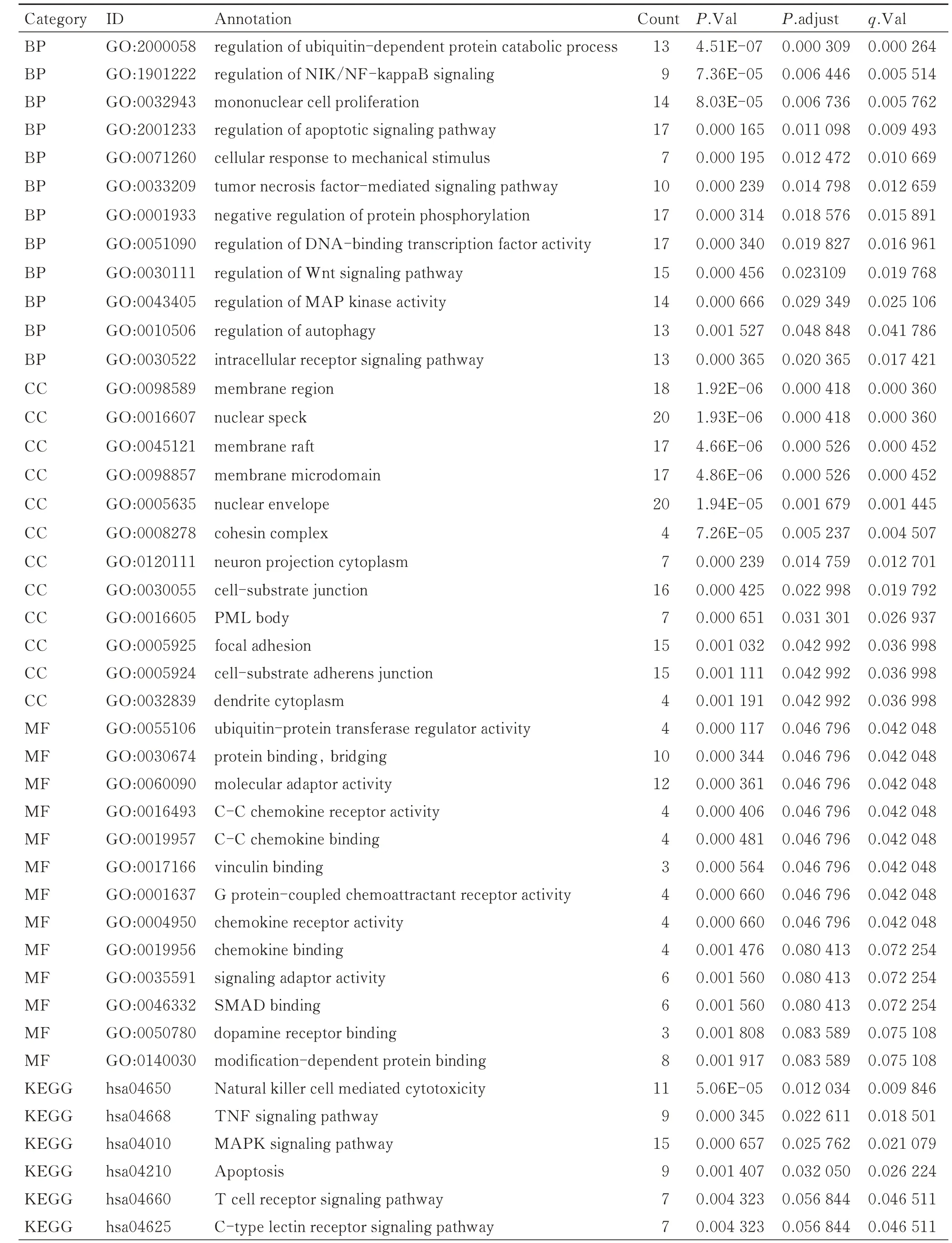
表2 GO 和KEGG 富集分析Tab.2 GO and KEGG enrichment analyses
2.5 PPI 网络分析及模块分析结果
PPI 网络图共有235 个节点,687 条互作关系,经MCODE 筛选出7 个集簇,展示得分较高的前4 个集簇,其中连接度最高的基因集簇,包括11 个节点,47条互作关系(图7)。然后使用cytohubba 插件采用MCC 和Degree 算法筛选出连结度最高的前10 个基因,再联合centiscape 插件中度(degree)、中介中心性(betweenness)和紧密度(closeness)等参数以及对应的P.adjust、Pvalue 和log2FC 值筛选核心基因,最后结合与OA 紧密相关的分子生物学意义综合分析确定8 个核心基因为:MAPK1、IL10、PTGS2、IL18、GSK3B、NFKBIA、TNFRSF1A、EGR1(图8 和表3)。

表3 核心基因关系网络的拓扑学分析Tab.3 Topological analysis of core gene relationship network

图7 PPI 网络及模块分析图Fig.7 PPI network and module analysis diagram
3 讨 论
OA 是中老年女性患者疼痛和致残的主要原因之一,不仅影响正常生活,也严重限制社交活动。目前以姑息治疗为主,尚无阻断或逆转关节退变的方法。随着疾病的不可逆性进展,多数患者需要接受终末期手术治疗。因此,及时诊治尤为重要。而血液作为传统的临床检测样本,因其近乎无创和易获得性,得到研究者的青睐。血液中PBMCs,是外周循环血中具有单个核细胞的总称,单个核是相对多个核或分叶核粒细胞而言,除间充质干细胞、造血干细胞和多潜能细胞等少数祖细胞群体外[6],主要包括淋巴细胞、单核细胞(Monocyte,Mo)、树突状细胞和NK 细胞等,Mo 可以发育为各种组织巨噬细胞,PBMCs为免疫系统的主要组成部分,参与人体免疫应答。近年来,人们开始关注OA 外周血成分的变化。PONCHEL等[7]发现,与健康人相比,OA 患者血中CD4+T 细胞和B 细胞更少,而CD8+T 细胞和记忆细胞更多,并且所表现的免疫功能障碍与衰老无关。临床上,王猛等[8]发现膝OA 患者外周血中性粒细胞-淋巴细胞比率和单核细胞-淋巴细胞比率明显高于健康志愿者,且对全膝置换术后关节功能评估有参考意义。
本研究基于GSE48556 数据集中所有女性血液样本,105 例髋或膝OA 患者和24 例正常对照,所有样本年龄和体质量指数基线指标无差异,二维和三维PCA 分析显示样本间距离小,可能与遗传背景接近有关,绝大部分OA 患者有疼痛,约一半服用非甾体类抗炎药。传统Log2FC 阈值筛选差异基因法没有考虑差异微弱的基因,可能会造成很大的偏倚,尤其对血中差异基因表达微弱疾病的鉴别。而GSEA 富集法是对所有表达水平的基因分析,将其与预定义的基因集比较,计算每个基因的富集分数,不仅可确定待测基因组在特定基因集中的表达状况及是否存在统计学差异,还可筛选出对富集得分贡献最大的基因子集。经过GSEA 富集筛选得到的3 120 个功能基因二次分析,发现很多功能基因没有统计学意义,log2FC值太小,以P.adjust<0.05 &|log2FC|>0.2 再次筛选,共得到目的基因292 个行GO、KEGG 和PPI 分析。
GO 分析显示,目的基因主要富集在“泛素依赖性蛋白质分解代谢过程的调节”、“NIK/NF-κB 的调节”、“单核细胞增殖”、“凋亡信号通路的调控”、“细胞对机械刺激的反应”、“TNF 介导的信号通路”、“Wnt信号通路的调控”、“MAP 激酶活性的调节”和“自噬的调节”等生物功能上。已有研究报道过OA 类似的病理机制。如LIU 等[9]认为泛素和泛素样修饰酶促机制在软骨细胞稳态方面发挥重要作用,包括调节细胞信号转导和蛋白质稳定性,处理细胞应激和炎症,以及维持软骨细胞分化和存活,可能成为治疗OA 新的靶点。王猛等[8]发现OA 患者外周血Mo 明显高于健康志愿者,说明单核细胞增殖与OA 有关,并且,外周血Mo 是滑膜巨噬细胞的来源细胞,而后者可能是OA 炎 症 的 发 起 者[10]。TER HEEGDE 等[11]研 究 发 现,机械负荷导致细胞应激反应和局部炎症,所诱发的疼痛与软骨损害的OA 样改变有关。NF-κB、凋亡信号通路、TNF 相关通路、Wnt 信号通路、MAP 激酶和自噬调节是常见的OA 病理机制,多与软骨细胞凋亡和滑膜细胞炎症有关,其中,NF-κB、TNF 相关通路和自噬与炎症机制密切相关,凋亡信号通路、Wnt 信号通路和MAP 激酶调节与软骨细胞凋亡更为紧密,两者之间又相互影响。KEGG 主要富集于以下4 条与OA 相关的通路:自然杀伤细胞介导的细胞毒性、TNF 信号通路、MAPK 信号通路和细胞凋亡。而关于TNF 信号通路、MAPK 信号通路和细胞凋亡在OA 病机中的研究较多,且在很多文献都有报道。关于“自然杀伤细胞介导的细胞毒性”,FENG 等[1]对GSE48556 数据集分析时也发现其是OA 差异基因主要富集通路之一,但目前尚未发现其参与OA 病机的相关实验研究。KEGG 富集分析进一步说明炎症和凋亡与OA 的病理变化密切相关。
通过PPI网络与centiscape插件筛选出8个核心基因:MAPK1、IL10、PTGS2、IL18、GSK3B、NFKBIA、TNFRSF1A、EGR1,其中仅MAPK1 和TNFRSF1A上调,其余基因都是下调。EGR1、PTGS2和IL18 都被之前的分析筛选为核心基因[1,4]。MAPK1 是MAP激酶家族一员,MAPK1 磷酸化加重软骨细胞肥大分化,在炎症因子共同作用下引起软骨细胞凋亡。IL10主要由单核细胞产生,具有强大的抗炎功能,限制炎症引起的过度组织破坏。CAMERON 等[12]在评估骨髓源性间充质干细胞(BM-MSC)和IL-10 在OA 中的组合作用的细胞实验中发现,IL-10 过表达可能会增强BM-MSC 介导的T 细胞抑制,但没有发现腺相关病毒-IL10 转导的BM-MSC 中炎性驱动软骨降解的显著调节。GSK3B 是糖原合酶激酶-3β,葡萄糖稳态的负调节剂,参与能量代谢、炎症、内质网应激和细胞凋亡等,HUANG 等[13]之前的研究中已将其筛选为核心基因,通过抑制WNT/β-catenin 通路维持软骨表型和软骨细胞外基质,拮抗软骨细胞凋亡。NFKBIA是NF-κB 抑制剂α,抑制参与炎症反应的NF-κB 复合物,TNFRSF1A 是肿瘤坏死因子受体超家族成员1A,与TNFα 受 体 结 合 抑 制 炎 症,XU 等[14]发 现,TNF-α 刺激下的软骨细胞NF-κB 通路高度富集,木兰素可以抑制NFKBIA 和TNFRSF1A 的表达而抑制OA 中软骨细胞的炎性反应。
然而,结合临床意义分析核心基因时发现:MAPK1、EGR1 和GSK3B 主要与软骨细胞凋亡有关,前两者正相关,后者负相关,但是正相关的EGR1表达量下调,与OA 软骨细胞凋亡增加不符,可能与机体自发的负反馈调节有关;IL18、PTGS2、IL10、NFKBIA 和TNFRSF1A 主要与炎症反应有关,前两者为促炎因子,后三者为抗炎因子,但是IL18 和PTGS2 的表达量都下调,可能与患者使用非甾体类抗炎药有关,也可能与机体自发保护反应有关。另外,本研究有以下不足:①对数据集的检索可能不完全,没有纳入全部相关资料;②没有建立合适的数学模型,仍然存在人为设置阈值,手工挑选目的基因;③没有进一步的实验验证。总之,本研究发现:①自然杀伤细胞介导的细胞毒性、TNF 信号通路、MAPK 信号通路、细胞凋亡等生物学过程与OA 病机密切相关;②MAPK1、IL10、PTGS2、IL18、GSK3B、NFKBIA、TNFRSF1A、EGR1 八个核心基因与OA炎症和细胞凋亡高度相关,可能是PBMCs 中区别于健康人的有效生物标志物,有望通过外周血检测识别OA。
