基于高效液相色谱指纹图谱和多成分含量的淫羊藿传统饮片、破壁粉与破壁饮片比较*
2022-05-16李友露缪艳燕曹国琼田洪星张永萍
李友露,徐 剑,2,3△,缪艳燕,2,3,曹国琼,2,3,田洪星,张永萍,2,3
(1. 贵州中医药大学药学院,贵州 贵阳 550025; 2. 贵州中药炮制与制剂工程技术研究中心,贵州 贵阳550025; 3. 国家苗药工程技术研究中心,贵州 贵阳 550025)
淫羊藿为小檗科植物淫羊藿Epimedium brevicomuMaxim.、箭叶淫羊藿Epimedium sagittatum(Sieb. etZucc.)Maxim.、柔毛淫羊藿Epimedium pubescensMaxim. 或朝鲜淫羊藿Epimedium koreanumNakai 的干燥叶,主产于贵州、湖北、四川、辽宁、陕西、湘西等地[1-2],主要活性成分为黄酮类成分。现代药理学研究表明,淫羊藿黄酮类成分具有抗氧化[3]、抗肿瘤[4]、抗炎[5]和抗骨质疏松[6]等作用,临床应用广泛[7]。超微粉碎技术因其破壁后与原传统饮片的物质基础一致,可加快有效成分溶出,提高生物利用度,且具有携带方便、用药灵活等优势,能解决市场需求量大、资源紧缺的问题[8-10]。淫羊藿破壁饮片是利用超微粉碎技术将传统饮片粉碎到粉体粒径(D90)小于45 μm 的破细胞壁粉体,再采用成型技术加工成40~80目的干燥颗粒状饮片。文献[1,11-12]表明,淫羊藿黄酮类成分易受生长环境、产地加工、炮制、储存等因素影响,可导致有效成分提取不均匀、稳定差等问题。但破壁技术也面临一系列问题,如经粉碎工艺后失去了原饮片的外观性状、显微结构特征,比表面积显著增大,以及破壁过程中的高速摩擦、滚动产生的热量对黄酮类成分产生影响,进而影响临床疗效[13-14]。本研究中采用高效液相色谱(HPLC)法建立了12 批淫羊藿传统饮片、破壁粉、破壁饮片的指纹图谱,并对其进行相似度评价,同时建立了5 个指标性成分的含量测定方法。现报道如下。
1 仪器与试药
1.1 仪器
XT-A400 型多功能粉碎机(永康市红太阳机电有限公司);HBM-109型超微粉碎机(瑞安市瀚博机电有限公司);GZX - 9240MBE 型电热鼓风干燥箱(上海博迅实业有限公司);ATY224 型电子分析天平(日本岛津公司,精度为100 mg);KQ-500E型超声波清洗器(昆山市超声仪器有限公司,功率为500 W,频率为40 kHz);1260 型Agilent 高效液相色谱仪(美国安捷伦公司);Milli-Q型超纯水仪(德国默克密理博公司)。
1.2 试药
朝藿定A 对照品(批号为MUST-14060312),朝藿定B 对照品(批号为MUST - 14062312),纯度均大于98%,购于成都曼思特生物科技有限公司;朝藿定C 对照品(批号为110756-200110),淫羊藿苷对照品(批号为110737-201516),宝藿苷Ⅰ对照品(批号为111852-201603),纯度均大于98%,购于中国食品药品检定研究院;水为自制超纯水;乙腈为色谱纯,其余试剂均为分析纯,购于国药集团化学试剂有限公司;淫羊藿饮片购自贵州同济堂药材有限公司,经贵州中医药大学何顺志教授鉴定为淫羊藿Epimedium brevicomuMaxim.、柔毛淫羊藿Epimedium pubescensMaxim.、箭叶淫羊藿Epimedium sagittatum(Sieb.et Zucc.)Maxim. 和朝鲜淫羊藿Epimedium koreanumNakai。详见表1。

表1 12批淫羊藿饮片信息Tab.1 Information of 12 batches of Epimedii Folium decoction pieces
2 方法与结果
2.1 色谱条件[15]
色谱柱:Odyssil C18柱(250 mm × 4.6 mm,5 μm);流动相:水(A)- 乙腈(B),梯度洗脱(0~10 min 时75%A~74%A,10~38 min 时74%A,38~43 min 时74%A~59%A,43~49 min时59%A~58%A,49~55 min时58%A,55~64 min 时58%A~35%A,64~85 min 时35%A~15%A);流速:1.0 mL/min;柱温:30 ℃;检测波长:270 nm;进样量:10 μL。
2.2 样品及溶液制备
2.2.1 样品制备
传统饮片:称取淫羊藿饮片适量,置多功能粉碎机中粉碎40 s,粉末过80目筛,密封,备用(编号为A1-A12)。
破壁粉:取上述细粉适量,使用超微粉碎机调整粗细旋转轴长度为0.6 cm,粉碎1 次,密封,备用(编号为B1-B12)。
破壁饮片:取上述破壁粉适量,混匀,加入15%乙醇,按料液比0.62∶1(m/V)制成软材,35目筛网制得湿颗粒,60 ℃干燥70 min,过筛整粒,密封,备用(编号为C1-C12)。
2.2.2 溶液制备
供试品溶液:取淫羊藿传统饮片、破壁粉、破壁饮片各0.2 g,精密称定,置具塞锥形瓶中,精密加入50%乙醇20 mL,密塞,称定质量,超声处理(频率为40 kHz,功率为500 W)45 min,冷却至室温,用50%乙醇补足减失的质量,摇匀,滤过,离心(转速为14000 r/min)10 min,取上清液,即得。
对照品溶液:取淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷Ⅰ对照品各适量,精密称定,置5 mL 容量瓶中,加甲醇制成每1 mL 分别含1.84,1.69,1.81,1.50,1.99 mg的溶液,摇匀,即得。
2.3 HPLC 指纹图谱建立
2.3.1 参照峰确定
本研究中所建立指纹图谱中淫羊藿苷的色谱峰分离良好,峰面积较大,保留时间适中,且为所有样品共有,参照2020年版《中国药典(一部)》淫羊藿项下淫羊藿苷的含量测定方法[15],以淫羊藿苷为参照峰。
2.3.2 方法学考察
精密度试验:取2.2.1 项下淫羊藿传统饮片(编号为A1)、破壁粉(编号为B1)和破壁饮片(编号为C1)各适量,精密称定,按2.2.2 项下方法制备供试品溶液,按2.1项下色谱条件连续进样测定6次,记录色谱图。结果共有峰相对保留时间的RSD分别为0.02%~0.14%、0.05%~0.09%、0.09%~0.18%(n=6),相对峰面积的RSD分别为1.44%~2.88%、2.46%~2.92%、1.57%~2.64%(n=6),表明仪器精密度良好。
重复性试验:取精密度试验项下供试品溶液,按2.1项下色谱条件分别进样测定6次,记录色谱图。结果共有峰相对保留时间的RSD为0.07%~0.15%、0.03%~0.16%、0.01%~0.24%(n=6),相对峰面积的RSD为 1.97%~2.70%、1.05%~2.82%、1.90%~2.86%(n=6),表明方法重复性良好。
稳定性试验:取精密度试验项下供试品溶液,按2.1 项下色谱条件分别于0,3,6,9,12,18,24 h 时进样测定,记录色谱图。结果共有峰相对保留时间的RSD为0.02%~1.27%、0.05%~1.35%、0.05%~1.29%(n=7),相对峰面积的RSD为1.74%~2.55%、1.44%~2.99%、1.20%~2.89%(n=7),表明供试品溶液在24 h 内稳定性良好。
2.3.3 指纹图谱建立及共有峰指认
取12 批淫羊藿传统饮片、破壁粉、破壁饮片,按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定,记录色谱图。将所得图谱录入中药色谱指纹图谱相似度评价系统(2012 版)软件,以中位数法分析,设置参照图谱。其中,传统饮片为编号A1-A12、破壁粉为编号B1- B12、破壁饮片为编号C1- C12,各自生成12批淫羊藿传统饮片、破壁粉、破壁饮片指纹图谱与对照指纹图谱叠加图(图1)。共标定13 个共有峰,结合对照品指认了其中的5 个共有峰,其中3 号峰为朝藿定A,4 号峰为朝藿定B,5 号峰为朝藿定C,7 号峰为淫羊藿苷,11号峰为宝藿苷Ⅰ。
2.3.4 相似度评价
12 批淫羊藿传统饮片、破壁粉、破壁饮片各自生成的共有模式图谱即为对照图谱(R),评价12批样品与各自对照图谱(R)之间的相似度,结果见表2 至表4。淫羊藿传统饮片为0.419~1.000,破壁粉为0.435~1.000,破壁饮片为0.425~1.000。12 批传统饮片A1- A4和A11,破壁粉B1-B3和B11,以及破壁饮片C1-C3和C11之间的相似度较高(>0.90);破壁粉(编号为B1-B3,B7,B10-B11)、壁破饮片(编号为C1-C3,C7,C10-C11)间的相似度较高,表现出近似的变化规律,表明淫羊藿种属的不同、产地或时间的不同导致其成分含量有所不同[1,3,16-17],质量稳定性有待提升。

表2 12批淫羊藿传统饮片指纹图谱相似度评价结果Tab.2 Evaluation results of fingerprint similarity of 12 batches of Epimedii Folium traditional decoction pieces
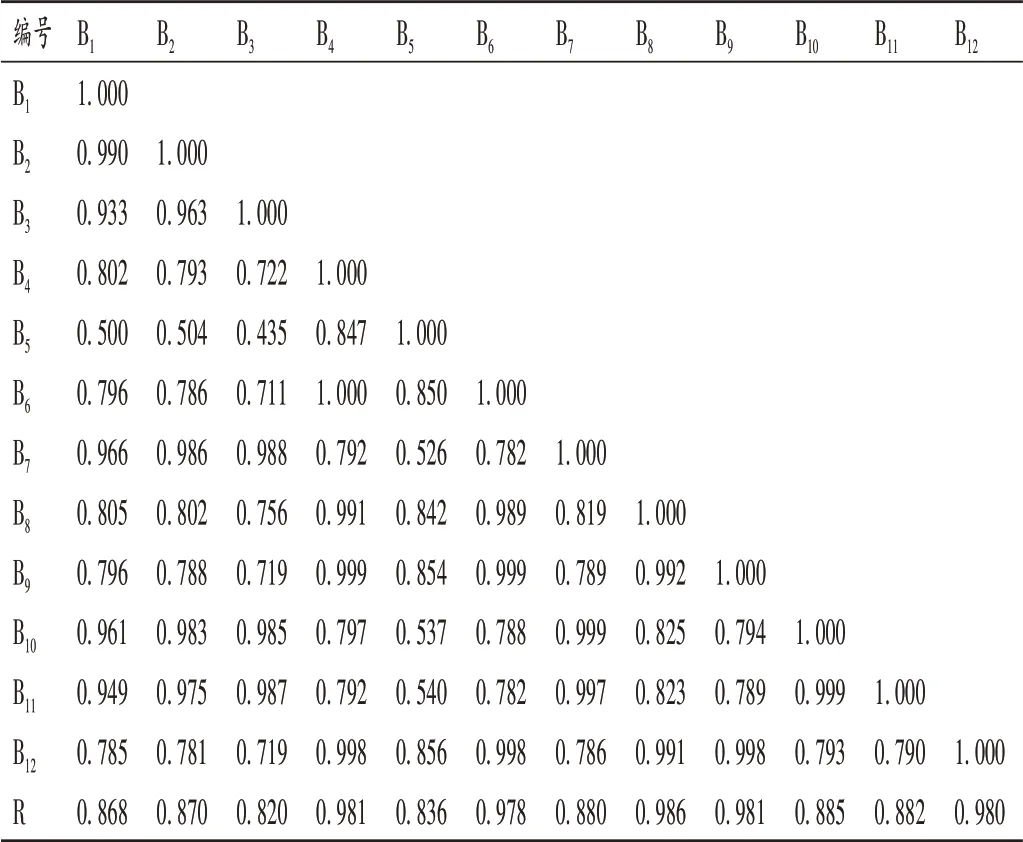
表3 12批淫羊藿破壁粉指纹图谱相似度评价结果Tab.3 Evaluation results of fingerprint similarity of 12 batches of Epimedii Folium cell-wall broken powder

表4 12批淫羊藿破壁饮片指纹图谱相似度评价结果Tab.4 Evaluation results of fingerprint similarity of 12 batches of Epimedii Folium cell-wall broken decoction pieces
2.3.5 指纹图谱相关性分析
将上述12 批淫羊藿传统饮片共有模式(S1)、破壁粉体共有模式(S2)、破壁饮片生成的图谱共有模式(S3)分别导入中药色谱指纹图谱相似度评价系统(2012版),生成对照图谱(R),进行相似度比较,结果见图2 和表5。结果表明,淫羊藿传统饮片、破壁粉及破壁饮片与对照图谱(R)之间的相似度较高(>0.99),表明淫羊藿传统饮片经破壁加工制成破壁粉、无添加成型工艺制备破壁饮片的过程中,有效成分的整体概貌没有发生明显变化,工艺流程质量稳定。
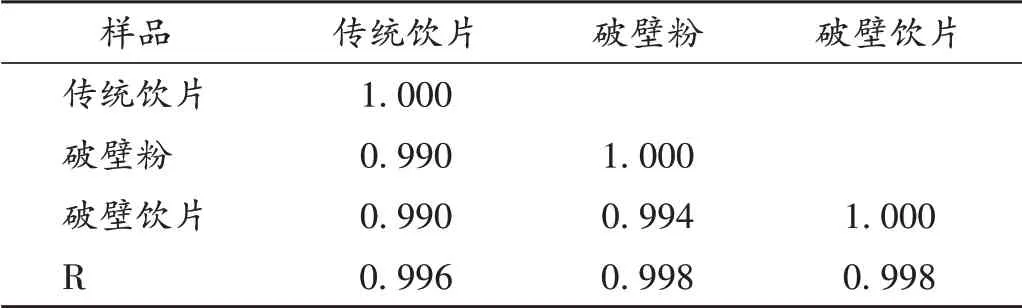
表5 12批淫羊藿传统饮片、破壁粉及破壁饮片相似度评价结果Tab.5 Similarity evaluation results of 12 batches of Epimedii Folium traditional decoction pieces,cell-wall broken powder and cell-wall broken decoction pieces
2.4 多指标成分含量测定
2.4.1 色谱条件与系统适用性试验
色谱条件同2.1 项下。取2.3.2 精密度试验项下供试品溶液(样品编号为A1)和2.2.2项下对照品溶液,按2.1 项下色谱条件分别进样10 μL 测定。色谱图见图3。结果朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ之间的分离度良好,相邻色谱峰间分离度均大于1.5,对样品的测定无干扰。
2.4.2 方法学考察
线性关系考察:精密吸取2.2.2 项下对照品溶液,加乙醇稀释成系列对照品溶液,朝藿定A质量浓度分别为1.84,0.92,0.59,0.30,0.15,0.07,0.04 mg/mL,朝藿定B 质量浓度分别为1.69,0.85,0.42,0.21,0.11,0.05,0.03 mg/ mL,朝藿定C 质量浓度分别为1.81,0.90,0.45,0.23,0.11,0.06,0.03 mg/ mL,淫羊藿苷质量浓度分别为1.50,0.75,0.38,0.18,0.09,0.05,0.02 mg/mL,宝藿苷Ⅰ分别为1.99,1.00,0.50,0.25,0.12,0.06,0.03 mg/ mL。按2.1 项下色谱条件进样测定,记录色谱峰峰面积,以质量浓度(X,mg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果见表6。

表6 线性关系考察结果(n=7)Tab.6 Results of the linear relation test(n=7)
精密度试验:精密吸取2.2.2 项下对照品溶液,按2.1项下色谱条件连续进样测定6次,测得朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积的RSD分别为0.24%,0.14%,0.12%,0.15%,0.48%(n= 6),表明仪器精密度良好。
重复性试验:取淫羊藿传统饮片(编号为A1)0.2 g,按2.2.2 项下方法平行制备供试品溶液6 份,按2.1 项下色谱条件进样测定6 次,测得朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积的RSD分别为0.98%,0.84%,0.52%,0.65%,1.14%(n= 6),表明方法重复性良好。
加样回收试验:取淫羊藿传统饮片(编号为A1)0.1 g,精密称定,共9 份,按80%,100%,120%的比例加入朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ对照品,按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定。结果平均加样回收率分别为101.01%,103.84%,99.25%,103.40%,102.65%,RSD分别为1.37%,0.95%,0.74%,0.81%,0.31%(n=9)。
2.5 多指标成分含量比较
通过对比淫羊藿传统饮片、破壁粉、破壁饮片的HPLC指纹图谱,发现其化学成分组成差异较小,但色谱峰峰面积存在一定差异。淫羊藿主要含有黄酮类成分,且朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ已纳入药典标准,上述成分是淫羊藿发挥药效的重要活性物质。本研究中采用HPLC法测定上述三者中多指标成分含量。
采用SPSS 26.0统计学软件对5个指标性成分进行多个独立样本非参数检验,P<0.05为差异有统计学意义。指标性成分总含量从高到低依次为破壁粉>破壁饮片>传统饮片分别为6.43%,5.76%,5.43%。由表7可知,淫羊藿破壁粉及破壁饮片各指标性成分平均含量均高于传统饮片,但均无显著差异(P>0.05)。
表7 12批淫羊藿传统饮片、破壁粉和破壁饮片多指标成分含量测定及非参数检验结果(±s,%)Tab.7 Content determination of multi-index components and nonparametric test results of 12 batches of Epimedii Folium traditional decoction pieces,cell-wall broken powder and cell-wall broken decoction pieces(±s,%)

表7 12批淫羊藿传统饮片、破壁粉和破壁饮片多指标成分含量测定及非参数检验结果(±s,%)Tab.7 Content determination of multi-index components and nonparametric test results of 12 batches of Epimedii Folium traditional decoction pieces,cell-wall broken powder and cell-wall broken decoction pieces(±s,%)
样品传统饮片破壁粉破壁饮片χ2值P值朝藿定A 0.62±0.150.65±0.150.67±0.100.4970.780朝藿定B 1.27±0.561.33±0.301.29±0.551.2720.529朝藿定C 1.94±2.242.55±2.552.06±1.670.2210.895淫羊藿苷1.46±0.251.75±0.471.57±0.303.4600.177宝藿苷Ⅰ0.14±0.050.15±0.050.17±0.042.5860.274
3 讨论
3.1 色谱条件优化
参照2020年版《中国药典(一部)》淫羊藿含量测定色谱条件,并进行优化,采用紫外-可见分光光度计对淫羊藿样品进行全波长扫描,270 nm 波长处检测信号较强,色谱峰数目较多,且分离度良好;曾比较色谱柱Agilent C18柱(250 mm×4.6 mm,5 μm),Diamonsil C18柱(250 mm × 4.6 mm,5 μm)和OdyssiL C18柱(250 mm ×4.6 mm,5 μm),OdyssiL C18柱(250 mm×4.6 mm,5 μm)的色谱峰分离效果较佳,峰形较好;曾比较乙腈- 水、乙腈-0.1%醋酸、乙腈-0.1%磷酸流动相系统,选择乙腈-水作为流动相,色谱峰数目较多、峰形和分离度均较好,曾比较柱温25,30,35 ℃,发现柱温过高出峰时间较快、分离效果较差,柱温太低出峰时间延长,故柱温选择30 ℃。最终确定2.1项下色谱条件。
3.2 样品提取条件优化
不同提取条件会影响有效成分的提取[18]。本研究中以共有峰峰面积/质量为评价指标,分别考察了提取方法(超声、回流、浸渍),提取溶剂(甲醇、无水乙醇、50%乙醇、80%乙醇),提取溶剂剂量(20,30,40 mL),提取时间(15,45,75 min)。结果发现,超声提取方便快捷,有利于节省时间,也能很好地把较多成分提取出来;提取溶剂为50%乙醇时色谱峰峰形和分离度均较好;提取45 min 可提取完全;提高溶剂剂量,共有峰峰面积/质量增加幅度不大。同时,考虑溶剂成本,最终选择提取溶剂剂量为20 mL。
3.3 不同批次淫羊藿传统饮片、破壁粉及破壁饮片化学组分传递分析
所建立淫羊藿传统饮片、破壁粉及破壁饮片的HPLC指纹图谱共标定13个共有峰,整体形貌上无化学组分数量、种类的变化,但其相似度波动范围较大(0.419~1.000,0.425~1.000,0.435~1.000),提示药材质量受品种、生长环境气候、产地等因素的影响较大。化学组分的差异即使采用相同工艺也会使中药成品质量不同,应加强把控淫羊藿药材的产地及加工方法。
为避免药材本身质量的不稳定性会放大制备流程中间体(破壁粉)、成品(破壁饮片)样品的差异性[19],根据前期得到的各样品的共有模式进行相关性分析,结果显示其相似度较高(>0.900),传统饮片经破壁粉碎技术及成型技术制成破壁饮片成品的整个制备流程中对化学组分的影响较小,成分传递好,与邓雯等[20]的研究结论一致。破壁过程中因高速滚动、摩擦,会产生大量的热,有可能造成淫羊藿主要含有的黄酮类成分分解、转化等,比较破壁前后淫羊藿黄酮类有效成分的含量之间的差异的研究鲜见报道。本研究结果表明,与传统饮片相比,破壁粉、破壁饮片淫羊藿黄酮类有效成分的提取率可得到提高,同时含量无明显差异。表明粉碎工艺的质量稳定、可靠,进一步提示淫羊藿利用该技术的可行性。
综上所述,本研究中从定性与定量2个角度对淫羊藿破壁饮片的黄酮类成分进行分析,为了更全面地理解破壁饮片制备过程的质量变化规律,后续仍可对其物理性质、生物利用度等进行进一步研究。
