肝豆状核变性合并肝细胞癌1例报告
2022-05-14罗绍轩
罗绍轩, 李 娅, 徐 峰
郑州大学第一附属医院 消化内科, 郑州 450052
1 病例资料
患者男性,32岁,自由职业者,因“右上腹隐痛1周”于2019年5月30日入本院。患者既往史、家族史无特殊,无吸烟史,偶饮酒。查体:慢性肝病面容,皮肤、巩膜无黄染,未见肝掌及蜘蛛痣,心肺听诊无异常。腹平坦,右上腹轻压痛,无反跳痛,脾脏肋缘下2 cm,肝脏未触及,移动性浊音阴性,双下肢无水肿。血常规:WBC 3.9×109/L,RBC 4.89×1012/L,Hb 151 g/L,PLT 99×109/L。凝血功能:PT 11.40 s,PTA 89.00%,国际标准化比值1.06。生化检验:ALT 53 U/L,AST 40 U/L,ALP 119 U/L,GGT 75 U/L,胆碱酯酶3.7 kU/L,TBil 5.4 μmol/L,DBil 3.1 μmol/L,IBil 2.3 μmol/L,总蛋白 67.8 g/L,Alb 45.1 g/L;血清铜蓝蛋白5 mg/dL(正常值15~45 mg/dL)。肿瘤标志物示:AFP 7.4 ng/mL,CEA、CA 19-9、CA125未见异常。传染病筛查(HBV血清学五项、HCV抗体、艾滋病毒抗体和梅毒螺旋体抗体)未见异常。2019年5月26日外院腹部超声示:(1)肝硬化伴肝内多发实性占位;(2)胆囊壁毛糙;(3)脾大。头颅CT未见明显异常,上腹部增强CT示:(1)肝脏右后叶占位,考虑肿瘤性病变可能性大;(2)肝硬化、肝内多发硬化结节;(3)脾大;(4)胆囊炎。外院CT于本院影像科会诊后考虑肝脏右后叶占位,肝细胞癌(HCC)可能性大。于本院完善腹部增强MRI示:(1)肝脏右后叶异常信号,可疑HCC(直径约10 mm);(2)肝硬化、肝内多发硬化结节;(3)脾大、门静脉高压;(4)肝门部及腹膜后多发小淋巴结影(图1)。肝纤维化无创诊断示:肝脂肪检测 CAP: 242 dB/m,肝硬度值20.5 kPa。
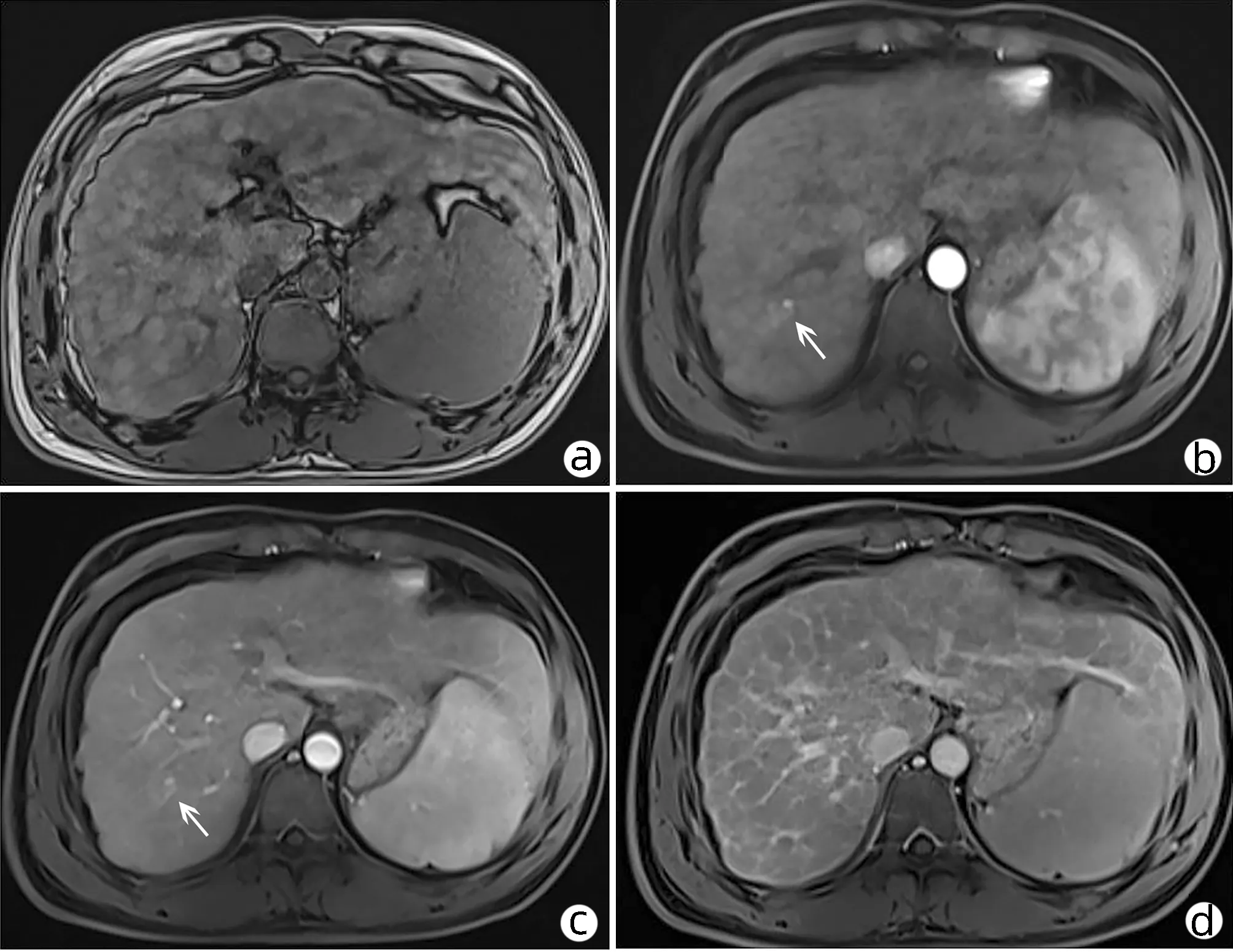
注:a,平扫期;b,动脉期(箭头示HCC);c,静脉期(箭头示HCC);d,延迟期。
综合临床表现及辅助检查结果,患者诊断为HCC、肝硬化(Child-Pugh A级)。但患者否认肝炎、酗酒、寄生虫感染及特殊用药史,肝硬化原因尚不明确,进一步完善相关检查。结果回示:甲、戊型肝炎标志物、自身免疫性肝病相关抗体(抗核抗体、抗线粒体抗体、抗可溶性肝抗原等)均为阴性。患者血清铜蓝蛋白水平较低,肝豆状核变性不能排除。继续完善24 h尿铜206.4 μg(正常<50 μg/d),眼科会诊示:于裂隙灯下可见角膜色素(Kayser-Fleischer,K-F)环,头颅MRI未见明显异常,肝豆状核变性基因检测示:ATP7B 基因c.2621C>T(p.Ala874Val)、c.2755C>G(p.Arg919Gly)复合杂合变异。基于以上结果,患者确诊为肝豆状核变性。肝硬化考虑由肝豆状核变性(肝型)导致。该患者1子1女均进行筛查,未患肝豆状核变性。
诊断明确后,给予低铜饮食,HCC予以行经肝动脉化疗栓塞术(TACE)。该患者青霉素皮试阴性,术后给予口服青霉胺起始剂量375 mg/d,无明显副反应后调整为750 mg/d治疗。现已规律随访24个月(图2),病情稳定。
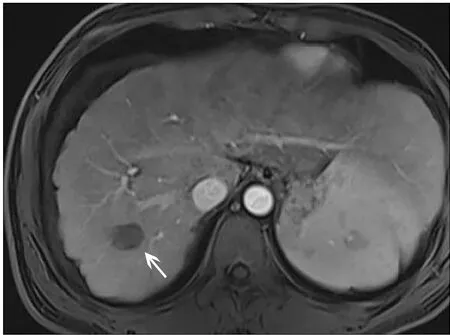
注:箭头示HCC TACE术后。
2 讨论
肝豆状核变性又称Wilson病(Wilson disease,WD),是一种常染色体隐性遗传的铜代谢异常疾病,人群患病率约为3∶100 000,致病基因携带率约1/90[1]。WD由位于13号染色体长臂的ATP7B基因突变引起,导致缺乏P型铜转运酶或功能下降,造成铜蓝蛋白合成障碍及铜离子转运减弱[2],未与铜蓝蛋白结合的“游离”铜被运送到血液,异常沉积在肝、脑、角膜、肾脏等组织,分别表现为转氨酶升高及肝硬化、神经精神症状、K-F环及肾功能损害等。WD发病多在5~35岁,在30岁以前以肝脏病变为主[3]。
已报道的ATP7B基因突变超过900种[4],大多为错义突变。在不同的区域和人群中,突变的分布具有较大差异。在亚洲及欧洲人群中最常见的突变分别为p.R778L和p.H1069Q,本例患者基因检测提示为复合杂合突变,其中一个突变位点位于Arg919Gly。Cheng等[5]报道其可能与WD脑型有关,但该患者未出现神经精神症状、头颅MRI未见代谢性脑病等特征性病变,考虑原因可能为基因表达受表观遗传和环境因素的共同影响。该患者仅ALT轻度升高,可能与肝脏储备功能较好有关。据统计,WD患者子女患病率约1/200[6],该患者子女均进行筛查未患有WD。
WD合并HCC较为罕见。在中国知网、万方以“肝豆状核变性”“Wilson病”“原发性肝癌”为关键词进行检索,同时在Pubmed以“hepatolenticular degeneration”“Wilson disease”“hepatocellular carcinoma”为关键词进行检索,共发现48例WD合并HCC患者,这些患者诊断HCC的平均年龄为44岁(10~73岁),男性比例为79%。最初认为铜沉积在肝脏有抑制HCC形成的作用[7-8],但近年越来越多的研究[9-13]表明,过多的铜可能通过产生活性氧而导致DNA双链断裂,同时刺激血管内皮生长因子及成纤维细胞生长因子2的表达,促进HCC的发生。Bruha等[14]对117例WD患者的研究显示,只有1例患者最终发展为HCC;一项对1186例WD患者进行多中心的回顾性分析[15]表明,最终8例患者进展为HCC,低于其他原因如病毒性肝炎、酒精性肝炎导致肝硬化并最终进展为HCC的年发生率[16]。考虑其原因可能为:(1)WD患者可能在进展为HCC之前,已经因肝硬化相关并发症死亡,而驱铜治疗延长了WD患者生存期,使得近年来WD合并HCC报道逐渐增多。van Meer等[17]报道其中1例WD患者随访50年后进展为HCC。(2)不明原因的肝硬化或者HCC患者,其原发病可能为WD,但因其发病率较低,临床医师认识不足,未能明确诊断[18]。
WD是少数可治但又具有致死性的遗传代谢病之一,国内外指南均推荐青霉胺作为一线驱铜治疗药物[3,19-20],但WD合并HCC的治疗方式目前尚未形成共识。既往文献报道的治疗方式包括外科手术切除、肝移植、肝动脉化疗栓塞、射频消融、全身化疗等,应根据患者及肿瘤情况选择合适的治疗方式。Kumagi等[21]报道1例WD合并HCC患者,采用TACE治疗后短期取得较好效果,但3年后因HCC复发引起肝衰竭而死亡。本例患者肝功能分级为Child-Pugh A级,肿瘤直径较小,肝损伤较轻,采用TACE微创治疗,定期随访24个月病情稳定,未出现肿瘤复发及肝衰竭等表现。表明TACE治疗肝功能储备较好的WD合并HCC患者近期疗效较好,远期疗效仍需要进一步随访明确。
本例患者以腹部隐痛就诊,血生化检验提示转氨酶轻度升高,但影像学检查已明确诊断为肝硬化、HCC,提示患者症状并不能预测病情的严重程度,值得临床重视。肝硬化原因不明确时,应警惕Wilson病的可能,及时完善血清铜蓝蛋白、血清铜、24 h尿铜及K-F环、头颅MRI等检查,必要时基因检测以明确诊断,早期给予驱铜等有效治疗,改善患者预后。
伦理学声明:本例报告已获得患者知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:罗绍轩负责课题设计,资料分析,撰写论文;李娅参与收集数据,修改论文;徐峰负责拟定写作思路,指导撰写文章并最后定稿。
