长链非编码RNA (LncRNA)在印度梨形孢促进大麦根部生长发育中的调控作用
2022-05-12郭楠楠刘天策胡心亭牛亚丹
郭楠楠 刘天策 史 硕 胡心亭 牛亚丹 李 亮
长链非编码RNA (LncRNA)在印度梨形孢促进大麦根部生长发育中的调控作用
郭楠楠 刘天策 史 硕 胡心亭 牛亚丹 李 亮*
河北工业大学化工学院, 天津 300130
印度梨形孢定殖植物会促进植物生物产量提高, 其分子机制有待深入挖掘。LncRNA是一类长链非编码RNA, 在植物生长发育过程中具有重要调控作用。然而, 目前我们还不清楚大麦中LncRNA是否对印度梨形孢的定殖有响应。本研究发现, 印度梨形孢定殖大麦会诱导大麦根系迅速发育, 促进较多根分枝。采用全基因组高通量测序RNA-seq和生物信息学方法鉴定LncRNA, 发现在定殖后3 d和7 d分别有752个和932个差异表达的LncRNA, 7 d相对于3 d有70个差异表达的LncRNA。其中在定殖后3 d有375个LncRNA表达上调, 377个LncRNA表达下调; 在定殖后7 d有459个LncRNA表达上调, 473个LncRNA表达下调; 7 d相对于3 d组中, 有39个LncRNA表达上调, 31个LncRNA表达下调。qPCR验证LncRNA的表达与RNA-seq结果一致。GO和KEGG分析表明, 在定殖大麦促生过程中, 部分LncRNA参与了激素信号途径的转录调控。该工作对于进一步理解LncRNA与靶基因的相互作用以及其对靶基因的调控功能提供了新的理论基础和实验依据, 并以LncRNA为靶点, 进行作物性状改良提供新的思路和方向。
LncRNA; 大麦; 印度梨形孢; RNA-seq; 转录因子; 细胞周期
研究人员曾经认为长链非编码RNA (LncRNA)是转录过程中产生的“无用物”, 最近发现其在植物的诸多调控过程中发挥重要作用[1]。尽管大多数LncRNA功能未知, 但已鉴定的LncRNA表现出组织特异性表达并参与蛋白定位以及调控相关疾病的特点[2-3]。顺式(或反式) LncRNA具有调节蛋白质活性或定位[4-5]的作用, 并可作为亚细胞结构的组织框架[2-3,6]。此外, 某些LncRNA可作为竞争性内源RNA (ceRNA)与小分子RNA (microRNA)配对[7-9]。
目前已对部分植物在遭受冷、热、干旱、盐和氮等一系列胁迫反应中的LncRNA进行了鉴定, 如毛果杨[10]、蒺藜苜蓿[11]、棉花[12]、玉米[13]和小麦[14-15]等植物。此外, 在对拟南芥的研究中发现, 冷诱导的LncRNA COOLAIR在春化早期起作用, 基因3′末端的反义转录机制可能是以条件/阶段依赖的方式调控基因转录, LncRNA COLDAIR可调节春化介导的表观遗传沉默从而应对冷胁迫[16-17]; LncRNA LDMAR在杂交稻中起着调节光周期敏感雄性不育的作用[18]; LncRNA DRIR能正向调控拟南芥对干旱和盐胁迫的响应[19]。另外, Zou等[20]和Karlik等[21]在大麦全基因组范围内发现了大约8000个LncRNA,并揭示了它们在过量硼处理下的表达模式。此外, 与LncRNA共表达编码转录本的功能注释揭示了离子转运、定位建立和叶对刺激响应的分子功能[22]。LncRNA HvCesA6调控大麦细胞壁的合成[23]。基于LncRNA-mRNA相互作用表明, LncRNA介导的调控丝氨酸/苏氨酸蛋白激酶SMG1信号转导途径可能与野生大麦XZ5的耐旱性有关[24]。Karlik等[25]报道了盐胁迫下大麦通过抑制LncRNAAK370814的表达反向促进基因表达, 并进一步明确了LncRNA-AK363461和LncRNAAAK370506在细胞中的定位。这些发现增进了我们对LncRNA作为抗旱重要调节因子的认识。然而, LncRNA在更广泛的环境条件下的生物学功能还需深入探索。
内生担子菌印度梨形孢()属蜡壳耳目, 梨形孢属[26]。与大麦、拟南芥、小麦、玉米、烟草、番茄[2,27-31]等多种植物形成了互惠菌根共生关系。具有促进植物生长、增强抗逆性和抗病性、促进营养物质和生物活性物质积累等多种功能[32-37]。分子生物学研究表明, 接种可诱导与激素信号转导、细胞壁代谢、碳水化合物运输代谢和根系形成等相关基因表达[38-39]。然而, 在促进大麦生长中, LncRNA的响应及调控机制仍未见报道, 需进行深入探索。
1 材料与方法
1.1 植株生长
将春大麦品种福大麦1号(162粒)用70%乙醇表面杀菌3 min, 然后在次氯酸钠(5%活性氯)中杀菌20 min。在无菌条件下, 用无菌水(pH 3.0)清洗1次, 无菌蒸馏水冲洗6次。将灭菌种子放在1/2 MS培养基的方形培养皿中, 每个培养皿中种6粒种子, 每个处理组(对照组即Mock、印度梨形孢定殖后3 d、印度梨形孢定殖后7 d)进行3个重复。在20℃的培养箱中培养, 条件如下: 光照8 h (荧光冷白, Toshiba FL40SSW/37, 180 µmol m–2s–1光子通量密度)/黑暗16 h, 22℃/18℃, 相对湿度60%。为了保证所有根系都能在培养基表面生长, 将含有种子的培养皿垂直放置。培养结束后, 挑选每个处理组长势良好的大麦各36颗, 对其根部进行取样, 样品送天津诺禾致源生物信息科技有限公司进行测序。
1.2 植物根系接种印度梨形孢
在CM培养基上生长3~4周, 可制备孢子悬浮液。在CM培养基中加入0.05%吐温-20的灭菌水, 用涂布棒轻轻刮擦平板表面, 收集孢子, 悬浮液通过miracloth (中国鼎国)过滤以除去菌丝体。将悬浮液在1359 ×下离心7 min收集孢子。用灭菌的吐温水清洗孢子3次。显微镜下, 血球计数板测定孢子密度。将孢子用无菌吐温水稀释浓度至105个孢子mL–1。接种时, 用移液枪将3 mL孢子悬浮液滴到方形培养皿中的植物根表面。
1.3 RNA提取及测序定量与鉴定
用TRIzol (Invitgen, #15596-018)法将对照组和处理组的根(0.5 g)在液氮中研磨成粉末。在1%琼脂糖凝胶上监测RNA降解和污染。分别使用NanoPhotometer分光光度计和Qubit 2.0 Flurometer中的Qubit RNA试剂盒检测RNA纯度与浓度。纯化的RNA用Revert-Aid第1链cDNA合成试剂盒进行反转录。根据制造商的说明, 使用Ribo-Zeror RNA Removal Kit (Epicentre, BioTechnoLogies)和NEB Next RULtraTMRNA Library Prep Kit for Illumina (New EngLand BioLabs)构建RNA-Seq文库。根据末端配对法, 使用Illumina HiSeq 2000对得到的文库进行测序。每个样本设置3个生物学重复。
1.4 差异表达基因的GO和KEGG富集分析
GO (Gene Ontology)是描述基因功能的综合性数据库, 可分为分子功能(molecular function)、生物过程(biological process)和细胞组成(cellular component) 3个部分。利用clusterProfiler R软件包与GO网站(http://geneontology.org/)对差异表达的LncRNA进行GO富集分析。KEGG (Kyoto Encyclopedia of Genes and Genomes)是一种数据库资源, 用于从分子水平信息, 尤其是基因组测序等高通量实验技术产生的大规模分子数据集(http://www.genome.jp/kegg/)了解生物系统(如细胞、生物体和生态系统)的高级功能和实用性。使用clusterProfiler R软件包测试KEGG通路中差异表达的LncRNA的统计富集情况。GO富集和KEGG通路富集以-value或adj小于0.05为显著富集。
1.5 实时定量聚合酶链反应(qRT-PCR)分析
分别用对照和处理组的总RNA (1 µg)与M-MLV Reverse转录酶来制备cDNA。经DNase I处理后, 将cDNA作为qRT-PCR的模板, 使用LncRNA特异性引物和靶mRNA特异性引物对所选择的LncRNA和mRNA进行定量分析。定量RT-PCR检测各基因的表达水平(SYBR绿色荧光法测定)。qPCR实验在Light Cyker96快速实时PCR系统(Roche)上进行, 该反应溶液含有2×ULtra SYBR 混合物10 µL, cDNA模板100 ng , 正反向引物10 µmol L–1。以HvUBIQUTIN为对照, 进行3次重复。扩增程序按以下步骤进行: 初始活化步骤在95℃下进行5 min, 然后进行30个循环(95℃ 20 s, 56℃ 35 s, 72℃ 35 s, 65℃ 20 s)。在每个循环结束时, 分别测定熔融曲线, 以保证单个PCR产物的扩增。
2 结果与分析
2.1 P. indica的定殖提高了大麦根的产量
大麦幼苗在处理的第3天和第7天的根长均比对照组显著增长(图1-A, B)。对3 d和7 d处理组与Mock组相比, 根长度分别增加了75%和58%。此外, 根在定殖7 d后分枝显著, 根数在3 d和7 d比Mock组分别增加了25%和42.85% (图1-C, D)。该结果表明,定殖能促进大麦根系生长和根数增加。
2.2 大麦根系对P.indica定殖反应的差异表达的LncRNA谱
为揭示大麦LncRNA对定殖的分子响应机制, 从12个大麦根部样本中提取RNA, 并进行全转录组序列测定。LncRNA鉴定的高通量测序流程和结果见图2。每个样本的RNA序列产生了1.042~1.285亿次raw reads和1.038~1.252亿次clean reads。Raw reads提交至NCBI (PRJNA71930)。利用Hisat将组装好的clean reads映射到大麦基因组上, 所有样本的平均映射率为74.13% (附表1)。利用RSeQC测定RNA-seq中LncRNA的饱和度见附表2。

图1 大麦在P. indica定殖后3 d和7 d形态、根长与根数统计结果图
A: 处理组与对照组在3 d的根长和根数, 标尺为 1.0 cm; B: 处理组与对照组在3 d根的分枝情况, 标尺为1.0 cm; C: 处理组与对照组在3 d和7 d的根长统计; D: 处理组与对照组在3 d和7 d的根数统计。进行3个生物学重复实验和统计分析。
A: root length and root number were compared between treatment and Mock at 3 days. Bar: 1.0 cm; B: branched roots were compared between treatment and Mock at 3 days. Bar: 1.0 cm; C: chart showing the statistical results of the root length of barley at 3 days and 7 days; D: chart showing the statistical results of the root number of barley at 3 days and 7 days.The experiments and statistical analysis were performed in three biological replicates.

图2 LncRNA鉴定的高通量测序流程图
层次聚类热图(图3)表明3个处理组(即3 d vs Mock、7 d vs Mock和7 d vs 3 d)的差异表达的LncRNA表达丰度不同, 说明这些基因位于不同的代谢途径及信号通路中。由图4可知, 在3 d vs Mock组中, 共有752个差异表达的LncRNA, 其中, 有375个LncRNA表达上调, 377个LncRNA表达下调; 在7 d vs Mock组中, 共有932个差异表达的LncRNA, 其中上调表达的LncRNA有459个, 下调表达的LncRNA有473个; 在7 d vs 3 d组中, 共有70个差异表达的LncRNA, 其中39个LncRNA表达上调, 31个LncRNA表达下调。
采用qRT-PCR对RNA-seq测序结果进行验证。使用特异性引物对随机选择的6个基因片段进行定量分析(表1)。结果表明, XLOC_070335、XLOC_318641和XLOC_067560这3个基因表达上调; XLOC_119623、XLOC_322519和XLOC_206343基因表达下调, 这6个基因片段的表达与测序分析结果一致(图5), 说明测序结果是可信的。
2.3 大麦根系对P. indica定殖反应的差异表达的LncRNA功能分析
在生物体内, 不同基因相互作用, 从而表现出生物学功能。通过pathway显著性富集可以探寻差异表达基因参与的最主要的生化代谢途径和信号转导途径。为了进一步研究这些LncRNA的功能, 我们对3个处理组中显著上调(附表3~附表5)和下调(附表6~附表8)的LncRNA进行了GO富集分析, 并绘制了GO富集柱状图(图6)。结果表明, 这些差异表达的LncRNA参与了生物学过程(BP)、细胞成分(CC)和分子功能(MF)。在3 d vs Mock组中对比显示, 在BP中, 差异表达的LncRNA在“代谢途径”和“单一生物代谢途径”中所占比例较高。在CC中, 占比高的部分为“细胞核”, 在MF中, 差异表达的LncRNA主要参与“催化活性”、“分子功能”和“结合”等过程。在7 d vs Mock组中对比显示, 在BP中, 差异表达的LncRNA在“代谢途径”、“单一生物代谢途径”与“蛋白质代谢途径”中所占比例较高, 在CC中, 占比高的部分为“细胞核”, 在MF中, 差异表达的LncRNA主要参与“催化活性”、“分子功能”、“结合”和“离子结合”等过程。在7 d vs 3 d组中对比显示, 在BP中, 差异表达的LncRNA在“防御反应”和“单一生物碳水化合物代谢途径”中所占比例较高, 在CC中, 统计到差异表达的LncRNA在“Smc5-Smc6络合物”、“U2型剪接体复合体”、“U2型mRNA释放后剪接体”、“mRNA释放后剪接体复合体”和“H/ACA RNP复合体”中富集, 在MF中, 差异表达的LncRNA主要参与“阳离子结合”与“过渡金属离子结合”等过程。

图3 差异表达的LncRNA的层次聚类热图
横坐标为样本名称, 纵坐标为差异表达的LncRNA, 左侧根据表达相似程度对基因进行聚类, 上方根据表达谱的相似程度对每个样本进行聚类, 由蓝至红表达量逐渐上调, 数字为均一化后的相对表达量。
The abscissa is the sample, the ordinate is the differentially expressed LncRNA. On the left side, the genes are clustered according to the degree of similarity of expression, and the upper part of the sample is clustered according to the degree of similarity of the expression profile. The relative expression level is gradually up-regulated from blue to red, and the number is the relative expression after standardization.

图4 差异表达LncRNA火山图
A: 3 d vs Mock比较组中差异表达的LncRNA; B: 7 d vs Mock 比较组中差异表达的LncRNA; C: 7 d vs 3 d比较组中差异表达的LncRNA。横坐标表示基因在不同样本或比较组合间的表达倍数变化(log2(Fold Change)), 横坐标的绝对值越大表明两个比较组合之间的表达变化倍数越大; 纵坐标表示表达差异的显著性水平。表达上调基因用红色点表示, 下调基因用绿色点表示, 蓝色点为未发生显著变化的基因(adj< 0.05) 。
A: volcano map of differentially expressed LncRNA in 3 days vs Mock comparison group; B: volcano map of differentially expressed LncRNA in 7 days vs Mock comparison group; C: volcano map of differentially expressed LncRNA in 7 days vs 3 days comparison group. The abscissa indicates the multiple change of gene expression between different samples or comparison combinations (log2(Fold Change)). The larger the absolute value of abscissa indicates the greater the multiple of expression change between the two comparison combinations; the ordinate indicates the significant level of expression difference. The up-regulated genes are represented by red dots, the down-regulated genes are represented by green dots, and the blue dots are genes that have not changed significantly (adj< 0.05).

表1 用于LncRNA差异表达鉴定的引物
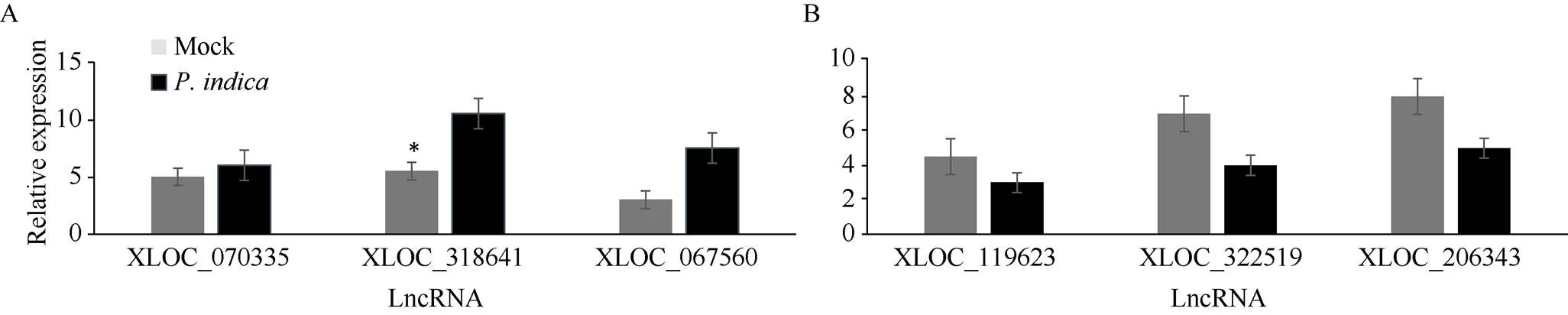
图5 qRT-PCR鉴定LncRNA的差异表达
A: LncRNA_XLOC_070335、XLOC_318641和XLOC_067560基因表达上调; B: LncRNA_XLOC_119623、XLOC_322519和XLOC_206343基因表达下调。qRT-PCR实验和数据分析进行3个生物学重复。*显著差异(< 0.05)。
A: LncRNA_XLOC_070335, XLOC_318641, and XLOC_067560 were up-regulated; B: LncRNA_XLOC_119623, XLOC_322519, and XLOC_206343 were down-regulated. The experiments of qRT-PCR and the data analysis were performed in three biological replicates. *:< 0.05.
KEGG通路分析采用KEGG API和R程序包Pathview进行。图7总结了3个对照组在KEGG途径中富集的差异表达的LncRNA。在3 d vs Mock组中, 上调基因在“酪氨酸代谢”、“次级代谢产物生物合成”、“氧化磷酸化”、“苯丙烷生物合成”、“苯丙氨酸代谢”途径中富集; 而下调基因在“次级代谢产物生物合成”、“苯丙烷生物合成”、“苯丙氨酸代谢”、“氧化磷酸化”途径中富集。在7 d vs Mock组中, 上调基因在“酪氨酸代谢”、“苯丙烷生物合成”、“苯丙氨酸代谢”、“氧化磷酸化”、“代谢途径”、“次级代谢产物生物合成”途径中富集; 下调基因在“苯丙烷生物合成”、“苯丙氨酸代谢”、“氧化磷酸化”、“代谢途径”、“次级代谢产物生物合成”途径中富集; 在7 d vs 3 d组中, 上调基因在“淀粉和蔗糖代谢”、“代谢途径”、“次级代谢产物生物合成”途径中富集; 下调基因在“代谢途径”、“次级代谢产物生物合成”途径中富集。
3 讨论
大麦是世界上种植最广泛、消费最多的粮食作物之一, 增加大麦产量是解决人口增长和气候变化问题的重要途径之一。参与植物代谢途径的相关LncRNA基因是影响产量的关键因素, 对植物适应环境胁迫具有重要调控作用。因此, 本研究对大麦进行全转录组测序旨在揭示LncRNA的潜在调控机制。作物与有益内生真菌的互作是一种自然而有效的增产方法。可以提高多种植物的生物产量, 并可诱导植物产生局部和系统抗性[28,36,40]。关于该真菌和植物的相互作用, 有从转录组进行的研究分析[41-42]。然而, 到目前为止, 还没有从LncRNA角度揭示对大麦的促生机制。
为了揭示定殖对大麦促进生长的分子机制, 我们进行了高通量RNA-seq分析。共鉴定出22,0761个LncRNA的转录本。通过KEGG分析植物激素信号转导途径, 发现参与生长素途径的基因(HORVU4Hr1G016160、HORVU2Hr1G109650、HORVU7Hr1G017790)表达上调(图8)。这与前期实验结果定殖促进了根系的形成与生长相吻合(图1), 而TCNS_00342393、TCNS_00211320、TCNS_00342393等LncRNA与生长素合成途径的基因均呈正相关; 此外, 编码DELLA蛋白的基因(HORVU2Hr1G016120、HORVU3Hr1G088780)表达下调。在赤霉素合成途径中, DELLA蛋白在信号转导中起着抑制子的作用, 这说明定殖能抑制DELLA 蛋白, 从而促进赤霉素合成途径。而LncRNA 如TCONS_00208317、TCONS_00237842、TCONS_00155869、TCONS_00555493均下调转录, 并与靶基因DELLA蛋白表达呈正相关。以上结果表明定殖大麦过程中, LncRNA参与了植物激素信号转导途径关键基因的表达调控, 且主要以正向调控为主。

图6 GO富集柱状图
A: 3 d vs Mock组中GO 的富集; B: 7 d vs Mock组中GO的富集; C: 7 d vs 3 d组中GO的富集; 横坐标表示GO条目名称, 分为3类(BP生物学过程, CC细胞组分, MF分子功能)用不同条框区分, 纵坐标为GO条目富集的基因数。
A: GO enrichment in 3 days vs Mock group; B: GO enrichment in 7 days vs Mock group; C: GO enrichment in 7 days vs 3 days group. The abscissa denotes the name of GO entry, which is divided into three categories by box (BP biological process, CC cell component, MF molecular function), distinguished by different frames, and the ordinate is the number of genes enriched by GO entry.

(图7)
图中纵坐标代表不同的通路, 横坐标代表相应通路显著差异表达基因占该通路所有基因的比例。圆圈大小代表富集在相应通路中的基因数目, 圆圈越大, 代表富集在该通路中的基因越多。颜色代表富集显著性, 越接近黑色, 代表越显著。
The ordinate represents different pathways, and the abscissa represents the proportion of differentially expressed genes in the corresponding pathway to all genes in the pathway. The circle size represents the number of genes enriched in the corresponding pathway, and the larger the circle, the more genes are enriched in the pathway. Color represents enrichment significance, and the closer it is to be black, the more significant it is.

图8 植物激素信号转导途径
4 结论
研究发现定殖后大麦根系生长加快。利用高通量RNA-seq和生物信息学分析和根部LncRNA全基因组鉴定表明在定殖后3 d和7 d分别有752个和932个差异表达的LncRNA, 7 d vs 3 d有70个差异表达的LncRNA。qPCR验证RNA-seq中关于LncRNA数据的有效性。GO及KEEG分析表明, 部分LncRNA参与了大麦根的形成和生长过程中激素信号途径的转录调控, 这些LncRNA可能通过反式作用调节靶基因, 也可能作为ceRNAs与某些miRNAs竞争。
附表 请见网络版: 1) 本刊网站http://zwxb.china crops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodicalzuowxb.aspx。
[1] Kitagawa M, Kitagawa K, Kotake Y, Niida H, Ohhata T. Cell cycle regulation by long non-coding RNAs., 2013, 70: 4785–4794.
[2] Wilusz J E, Sunwoo H, Spector D L. Long noncoding RNAs: functional surprises from the RNA world., 2009, 23: 1494–1504.
[3] Rinn J L, Chang H Y. Genome regulation by long noncoding RNAs., 2012, 81: 145–166.
[4] Kang C, Liu Z. Global identification and analysis of long non-coding RNAs in diploid strawberryduring flower and fruit development., 2015, 16: 815.
[5] Li C, Qiao Z, Qi W, Wang Q, Yuan Y, Yang X, Tang Y, Mei B, Lyu Y, Zhao H, Xiao H, Song R. Genome-wide characterization of cis-acting DNA targets reveals the transcriptional regulatory framework ofin maize., 2015, 27: 532–545.
[6] Chekanova J A. Long non-coding RNAs and their functions in plants., 2015, 27: 207–216.
[7] Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi P P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?, 2011, 146: 353–358.
[8] Rubio-Somoza I, Weigel D, Franco-Zorilla J M, Garcia J A, Paz-Ares J. ceRNAs: miRNA target mimic mimics., 2011, 147: 1431–1432.
[9] Fan C, Hao Z, Yan J, Li G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize., 2015, 16: 793.
[10] Shuai P, Liang D, Tang S, Zhang Z, Ye C Y, Su Y, Xia X, Yin W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in., 2014, 65: 4975–4983.
[11] Wang T Z, Liu M, Zhao M G, Chen R, Zhang W H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress inusing genome-wide high-throughput sequencing., 2015, 15: 131.
[12] Lu X, Chen X, Mu M, Wang J, Wang X, Wang D, Yin Z, Fan W, Wang S, Guo L, Ye W. Genome-wide analysis of long noncoding RNAs and their responses to drought stress in cotton (L.)., 2016, 11: e0156723.
[13] Lyu Y, Liang Z, Ge M, Qi W, Zhang T, Lin F, Peng Z, Zhao H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (L.)., 2016, 17: 350.
[14] Xin M, Wang Y, Yao Y, Song N, Hu Z, Qin D, Xie C, Peng H, Ni Z, Sun Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing., 2011, 11: 61.
[15] Shumayla, Sharma S, Taneja M, Tyagi S, Singh K, Upadhyay S K. Survey of High Throughput RNA-Seq Data reveals potential roles for LncRNAs during development and stress response in bread wheat., 2017, 8: 1019.
[16] Heo J B, Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA., 2011, 331: 76–79.
[17] Swiezewski S, Liu F, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of anPolycomb target., 2009, 462: 799–802.
[18] Ding J, Lu Q, Ouyang Y, Mao H, Zhang P, Yao J, Xu C, Li X, Xiao J, Zhang Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice., 2012, 109: 2654–2659.
[19] Qin T, Zhao H, Cui P, Albesher N, Xiong L. A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance., 2017, 175: 1321–1336.
[20] Zou Y, Tang H, Li T, Sun M, Qu X, Zhou J, Yang C, Mu Y, Jiang Q, Liu Y, Chen G, Chen G, Zheng Y, Wei Y, Lan X, Ma J. Identification and characterization of mRNAs and LncRNAs of a barley shrunken endosperm mutant using RNA-seq., 2020, 148: 55–68.
[21] Karlik E, Gözükırmızı N. Evaluation of barley LncRNAs expression analysis in salinity stress., 2018, 54: 198–204.
[22] Unver T, Tombuloglu H. Barley long non-coding RNAs (LncRNA) responsive to excess boron., 2020, 112: 1947–1955.
[23] Michael A H, Bryan P, Amanda S B, Sarah A K, Wei Y, Steven R S, Nicholas C C. Small-interfering RNAs from natural antisense transcripts derived from a cellulose synthase gene modulate cell wall biosynthesis in barley., 2008, 105: 20534–20539.
[24] Qiu C W, Zhao J, Chen Q, Wu F. Genome-wide characterization of drought stress responsive long non-coding RNAs in Tibetan wild barley., 2019, 164: 124–134.
[25] Karlik E, Gozukirmizi N. Expression analysis of LncRNA AK370814 involved in the barley vitamin B6 salvage pathway under salinity., 2018, 45: 1597–1609.
[26] Weiss M, Selosse M A, Rexer K H, Urban A, Oberwinkler F. Sebacinales: a hitherto overlooked cosm of heterobasidiomycetes with a broad mycorrhizal potential., 2004, 108: 1003–1010.
[27] Ghaffari M R, Mirzaei M, Ghabooli M, Khatabi B, Wu Y, Zabet-Moghaddam M, Mohammadi-Nejad G, Haynes P A, Hajirezaei M R, Sepehri M, Salekdeh G H. Root endophytic fungusimproves drought stress adaptation in barley by metabolic and proteomic reprogramming., 2019, 157: 197–210.
[28] Sherameti I, Venus Y, Drzewiecki C, Tripathi S, Dan V M, Nitz I, Varma A, Grundler F M, Oelmuller R. PYK10, a b-glucosidase located in the endoplasmatic reticulum, is crucial for the beneficial interaction betweenand the endophytic fungus., 2008, 54: 428–439.
[29] Zhang W, Wang J, Xu L, Wang A, Huang L, Du H, Qiu L, Oelmuller R. Drought stress responses in maize are diminished by., 2018, 13: e1414121.
[30] Abdelaziz M E, Abdelsattar M, Abdeldaym E A, Atia M A M, Mahmoud A W M, Saad M M, Hirt H.alters Na+/K+homeostasis, antioxidant enzymes and LeNHX1 expression of greenhouse tomato grown under salt stress.(Amsterdam), 2019, 256: 108532.
[31] Varma A, Verma S, Sudha X, Sahay N, Butehorn B, Franken P., a cultivable plant-growth-promoting root endophyte., 1999, 65: 2741–2744.
[32] Yang L, Cao J L, Zou Y N, Wu Q S, Kuča K.: a root endophytic fungus and its roles in plants., 2020, 48: 1–13.
[33] Bagde U S, Prasad R, Varma A. Impact of culture filtrate ofon biomass and biosynthesis of active ingredient aristolochic acid inMart., 2013, 6: 29–37.
[34] Kumar V, Sarma M V, Saharan, Srivastava R, Kumar L, Sahai V, Bisaria V S, Sharma A K. Effect of formulated root endophytic fungusand plant growth promoting rhizobacteria fluorescent pseudomonads R62 and R81 on Vigna mungo., 2012, 28: 595–603.
[35] Sharma G, Agrawal V. Marked enhancement in the artemisinin content and biomass productivity inL. shoots co-cultivated with., 2013, 29: 1133–1138.
[36] Vadassery J, Ranf S, Drzewiecki C, Mithofer A, Mazars C, Scheel D, Lee J, Oelmuller R. A cell wall extract from the endophytic funguspromotes growth ofseedlings and induces intracellular calcium elevation in roots., 2009, 59: 193–206.
[37] Yadav V, Kumar M, Deep D K, Kumar H, Sharma R, Tripathi T, Tuteja N, Saxena A K, Johri A K. Withdrawal: a phosphate transporter from the root endophytic fungusplays a role in phosphate transport to the host plant., 2010, 285: 26532–26544.
[38] Lee Y C, Johnson J M, Chien C T, Sun C, Cai D, Lou B, Oelmüller R, Yeh K W. Growth promotion of Chinese cabbage andbyis not stimulated by Mycelium-synthesized auxin., 2011, 24: 421–443.
[39] Sun C, Johnson J M, Cai D, Sherameti I, Oelmuller R, Lou B.confers drought tolerance in Chinese cabbage leaves by stimulating antioxidant enzymes, the expression of drought-related genes and the plastid-localized CAS protein., 2010, 167: 1009–1017.
[40] Baltruschat H, Fodor J, Harrach B D, Niemczyk E, Barna B, Gullner G, Janeczko A, Kogel K H, Schafer P, Schwarczinger I, Zuccaro A, Skoczowski A. Salt tolerance of barley induced by the root endophyteis associated with a strong increase in antioxidants., 2008, 180: 501–510.
[41] Schafer P, Pfiffi S, Voll L M, Zajic D, Chandler P M, Waller F, Scholz U, Pons-Kuhnemann J, Sonnewald S, Sonnewald U, Kogel K H. Manipulation of plant innate immunity and gibberellin as factor of compatibility in the mutualistic association of barley roots with., 2009, 59: 461–474.
[42] Zuccaro A, Lahrmann U, Guldener U, Langen G, Pfiffi S, Biedenkopf D, Wong P, Samans B, Grimm C, Basiewicz M, Murat C, Martin F, Kogel K H. Endophytic life strategies decoded by genome and transcriptome analyses of the mutualistic root symbiont., 2011, 7: e1002290.
Regulation of long non-coding RNA (LncRNA) in barley roots in response tocolonization
GUO Nan-Nan, LIU Tian-Ce, SHI Shuo, HU Xin-Ting, NIU Ya-Dan, and LI Liang*
College of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, China
The molecular mechanism of biomass enhancement byin colonization plants needs to be further explored. LncRNA is a kind of long-chain non-coding RNA, which plays an important role in the regulation of plant growth and development. However, it remains unclear whether barley LncRNAs are responsive tocolonization. It was found that barley roots exhibited fast development and large roots branched aftercolonization. Genome-wide high throughput RNA-seq and bioinformatical analysis showed that 752 and 932 differentially expressed LncRNAs were detected in responsive toat 3-day and 7-day after colonization, respectively. And 70 differentially expressed LncRNAs were found at 7-day compared to 3-day. Among these, 375 were up-regulated and 377 were down-regulated after 3 days’ colonization, and 459 were up-regulated and 473 were down-regulated after 7 days’ colonization, 39 were up-regulated and 31 were down-regulated in 7-day to 3-day comparison group. The qPCR results verified the validity of LncRNAs data in RNA-seq. GO and KEGG analysis indicated that a few LncRNAs might be involved in the molecular functions, cellular components, and biological processes uponcolonization. This study provides a new theoretical basis and experimental basis for further understanding of the interaction between LncRNAs and coding sequences and regulatory functional networks, and provides new ideas and directions for crop shape improvement based on LncRNAs.
LncRNA; barley;; RNA-seq; transcription factor; cell cycle
2021-04-13;
2021-10-19;
2021-11-02.
10.3724/SP.J.1006.2022.11043
通信作者(Corresponding author):李亮, E-mail: liangli@hebut.edu.cn
E-mail: 15033355861@163.com
本研究由国家自然科学基金项目(31801948), 河北省重点研发计划项目(19226505D)和河北省自然科学基金项目(C2021202005)资助。
This study was supported by National Natural Science Foundation of China (31801948), the Key Research & Development Projects in Hebei Province (19226505D), and the Natural Science Foundation of Hebei Province (C2021202005).
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20211101.1150.006.html
