利用BSA-Seq方法快速定位作物农艺性状QTL/基因概述
2022-05-10周文期刘忠祥王晓娟何海军周玉乾杨彦忠连晓荣李永生
周文期 刘忠祥 王晓娟 何海军 周玉乾 杨彦忠 连晓荣 李永生


摘要:为了利用生物学技术和手段来解决农业问题,让基因组测序技术、转基因及基因编辑等现代分子生物学技术为农业遗传育种服务,实现常规育种与分子设计育种的紧密结合,加速作物精准育种进程,近年颇受学术界关注。现梳理了一些分子生物学专用名词概念,概述了目前在正向遗传学研究中如何利用极端表型材料基于全基因组测序的BSA-Seq方法(MutMap法、MutMap+法、MutMap-Gap法、QTL-seq法)快速定位重要农艺性状的QTL/基因,为快速定位和克隆候选基因提供新思路。
关键词:QTL定位; BSA-Seq方法;遗传群体;基因克隆;农艺性状;分子育种
中图分类号:S336;Q78 文献标志码:A 文章编号:1001-1463(2022)04-0001-10
doi:10.3969/j.issn.1001-1463.2022.04.001
Rapid Mapping of QTL/gene for Agronomic Traits in Crops Using BSA-Seq Method
ZHOU Wenqi, LIU Zhongxiang, WANG Xiaojuan, HE Haijun, ZHOU Yuqian, YANG Yanzhong, LIAN Xiaorong, LI Yongsheng
(Institute of Crops, Gansu Academy of Agricultural Sciences, Lanzhou Gansu 730070, China)
Abstract:In order to resolve the agriculture problem using biological technologies and tools, modern molecular biology technologies such as genome sequencing, transgenic technology and gene editing could serve agricultural genetics and breeding, to realize the close intergration of traditional breeding and molecular design breeding, and accelerate the process of precision breeding of crops, which has attracted academic attention in recent years. In this study, we sorted out some molecular biological terminology concepts, and summarized how to rapidly locate QTL/gene for important agronomic traits using extreme phenotypic materials and BSA-Seq (Bulked segregate analysis-sequencing) method (MutMap, MutMap+, MutMap-Gap and QTL-seq method) based on the whole genome sequencing in forward genetics, providing new ideas for rapid mapping and cloning of candidate genes.
Key words:QTL mapping;BSA-seqmethod;Genetical population;Map-based cloning;Agronomic traits;Molecular breeding
過去人们依靠传统育种方法获取农作物新品种,根据所需新品种的特性,轮回选择并纯化其亲本,然后通过杂交、回交或直系筛选程序来完成。传统育种在整个农业发展中发挥了至关重要的作用,但同时也存在着育种上的盲目性、经验性、长周期和不确定因素等诸多问题。目前,遗传育种已经历了从原始驯化选育阶段由耕作者选育具有广泛表型变异的地方农家品种(育种1.0版);常规育种阶段由职业育种家通过预先设计杂交育种试验选育现代栽培品种(育种2.0版);进入依靠生物技术的分子育种阶段,采用分子标记辅助选择及转基因技术的手段实现对品种单一目标性状的导入与修饰(育种3.0版)。随着基因工程和现代信息技术的迅速崛起,育种也逐渐步入互联网+大数据+人工智能“三位一体”的设计育种时代,利用分子生物学技术和遗传学方法控制动植物群体数量和质量均得以实现,从而将全面改写生物育种的策略和方法,推动育种技术向智能化阶段(育种4.0版)发展[1 - 2 ]。生物技术向人类展示了巨大发展潜力,创造着农业革命的未来。利用分子设计育种彰显出比传统育种更为突出的优越性,结合基因组学、表型组学、蛋白组学及代谢组学等多组学数据进行生物信息学的整合和分析、优化和筛选,获取最佳育种目标基因型,通过转基因技术或基因编辑手段,可高效精准地培育出目标新品种,缩短育种周期2~5 a,提高育种效率,因此智能化育种是未来作物育种的必然选择,其精准性、科学性和高效性都将带领作物育种进入一个新的时代[2 - 3 ]。智能化育种或分子设计育种4.0版的关键方案就是如何合理设计具有理想株型(玉米、小麦和水稻等有各自不同的理想株型,如玉米合理株高、穗上叶夹角减小、增强耐密性、株型性状之间的协同调控机制等)、高产、高抗、绿色、优质等综合农艺性状的优良品种。为了实现设计目标,定位和克隆控制农艺性状的关键基因,及解析其遗传调控机理至关重要[4 - 7 ]。通过正向遗传方法的图位克隆技术是传统经典的基因克隆方法,但需要构建永久遗传群体或F2 ∶ 3分离群体,周期很长,存在高密度分子标记筛选工作量大、标记难以获得等缺点。我们通过介绍如何利用作物极端表型材料基于混合分组测序(Bulked segregate analysis-sequencing BSA-Seq)的MutMap方法快速获得重要农艺性状的QTL/基因,旨在为定位和克隆候选基因提供思路和方法。
1 分子生物学一些专用名词介绍
1.1 基因测序及克隆相关概念
1.1.1 表型与基因型(Phenotype andgenotype) 又称性状,是基因型(Genetype)和环境(Environment)共同作用的结果,即P=G+E;基因型是指某一生物个体全部基因组合的总称[8 ]。
1.1.2 质量性状(Qualitativecharacter) 指属性性状,是指同一种性状的不同表现型之间不存在连续性的数量变化,而呈现质的中断性变化的那些性状,组间差异显著。质量性状通常受一个或少数几个主效基因调控,不易受环境因素影响。比如花色、抗病性、抗虫性、育性等表现为质量性状遗传的特点[8 ]。
1.1.3 数量性状(Quantitative character) 指某一性状在群体中变异呈连续性正态分布,无法明确分组,如动植物的高度、长度、成熟期、产量等经济特性,这类性状无法通過表型来推断其基因型,易受环境影响和遗传背景影响[8 ]。
1.1.4 数量性状基因座(Quantitative trait locus,QTL) 指控制数量性状的基因在基因组中的位置。把能控制数量性状的位点就叫做QTL[8 ]。
1.1.5 遗传标记(Genetic marker) 指可追踪染色体、染色体某一片段或某个基因座在家系中传递的任何一种遗传特性。具有两个基本特征,即可遗传性和可识别性。某种生物的任何有差异表型的基因突变型均可作为遗传标记[8 ]。
1.1.6 遗传连锁图谱(Genetic linkage map) 指基因或DNA标记在染色体上的相对位置与遗传距离。单位由基因或DNA片段在染色体交换过程中分离的频率厘摩(cM)来表示。1 cM表示每次减数分裂的重组频率为1%[8 ]。
1.1.7 图位克隆(Map-based cloning) 又称定位克隆,1986年首先由剑桥大学的Alan coulson提出[9 ],可用于分离大部分性状的相关基因,它是通过分析突变位点与已知分子标记的连锁关系来确定候选基因,根据目的基因在染色体上的位置进行定位[8 ]。群体中的个体在某一表型性状上有遗传差异,导致这些差异的基因可以被已知的分子标记定位在特定染色体位置。随着各种分子标记技术的发展和高密度连锁图谱构建完成,图位克隆技术已经成为分离生物基因最常规的手段之一。
1.1.8 全基因组重测序(Genome-wide resequencing)
全基因组重测序是对基因组序列已知的个体进行基因组水平测序,并在个体或群体水平上进行差异性分析的方法。如拟南芥两个已测序亚种Columbia和Landsberg、水稻粳稻和籼稻两个亚种代表日本晴和9311、玉米Reid 和Lancaster类群代表自交系B73和Mo17等,均已经有参考的基因组序列。对有优异农艺性状的个体或者群体进行基因组重测序,将结果与已有的基因序列进行比对分析,找出控制重要农业性状的QTLs或目标基因,为育种精准控制表型所用[8 ]。
1.1.9 简化基因组测序(Reduced-representation genome sequencing,RRGS) 指利用限制性内切酶分割基因组DNA,选择性地回收一定长度范围内的酶切片段进行高通量测序,获得海量遗传多态性标签序列来充分代表目标物种全基因组信息的测序策略[8 ]。
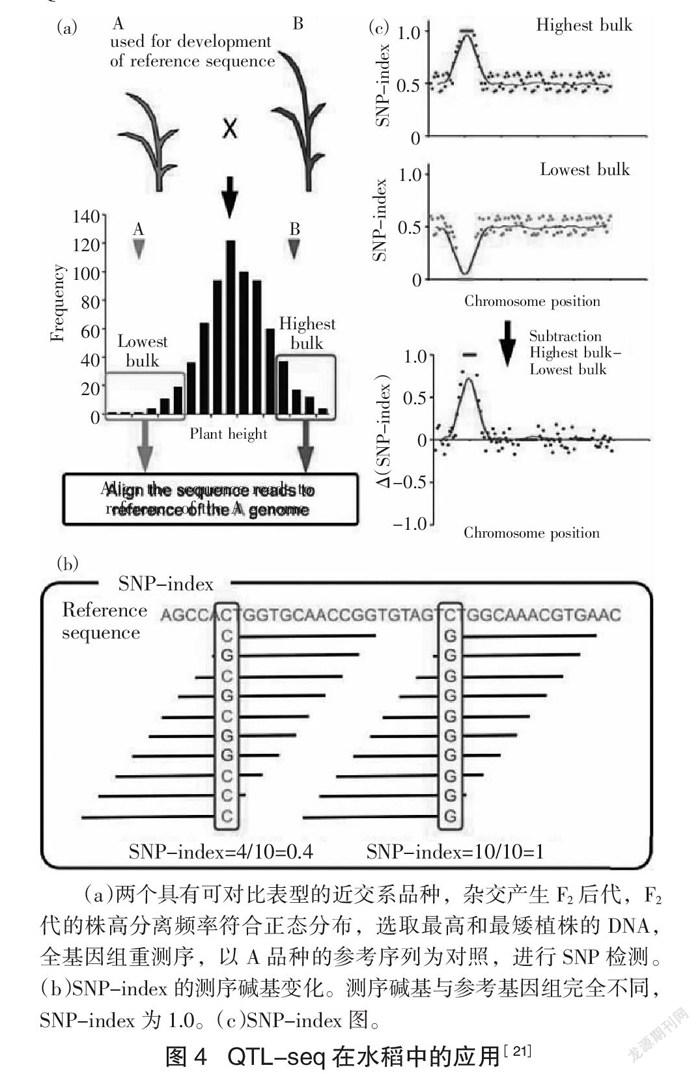
1.1.10 混合分组测序分析法(Bulked segregate analysis-sequencing,BSA-Seq) 又称分离体分组混合分析法或集团分离分析法[10 ],常用于单个QTL、突变位点定位等。将目标性状在F2后代或RILs群体中的极端表型,如株高或株矮、叶宽或叶窄、抗逆性状表现高抗或敏感等中2组个体的DNA分别混合成2个DNA池,测序比较两组群体在多态位点(SNPs)的等位基因频率(AF)是否具有显著差异,然后利用高密度分子标记在两池中进行标记与性状间的共分离分析,通过SNP值计算,筛选与性状相关联的QTL/基因[8 ]。
1.1.11 RNA的混合分组测序法(Bulked segregant RNA-sequencing,BSR-Seq) 指将转录组测序与集群分离分析相结合,在转录组范围内开发SNPs,筛选与性状紧密连锁的SNPs,进行功能基因的定位,同时进行基因差异表达分析等转录组常规分析的技术。基于RNA表达水平的BSR,更多的是转录本的比对[8 ]。
1.1.12 全基因组关联分析(Genome-wide association study,GWAS) 是一种对全基因组范围内的常见遗传变异(单核苷酸多态性和拷贝数)多态性总体关联分析的方法[11 ],在全基因组范围内进行整体研究,能够一次性对目标性状进行轮廓性概览,适用于复杂性状的研究[8 ]。
1.2 遗传群体定义及构建方法
1.2.1 作图群体(Mapping population) 指双亲通过自交和杂交产生后代,后代中性状和标记位点基因型均是分离的群体,也叫分离群体,常被用于遗传图谱构建,因此也叫作图群体[8 ]。作图群体被分为初级作图群体和次级作图群体2种。初级作图群体根据遗传稳定性又可分为临时性分离群体(F2、F3、BC1)和永久性分离群体(单倍体群体、重组自交系群体、永久性F2分离群体IF2等),个体间遗传背景差异较大。次级作图群体的群体基因组背景高度一致且与轮回亲本相同,个体仅含少量供体亲本片段,主要包括近等基因系类群体(NILs)、单片段代换系(SSSL)群体和染色体片段置换系(CSSL)群体。
1.2.2 重组自交系(Recombinant inbred line,RIL)
群体是杂种后代经过多代自交而产生的一种作图群体,即重组近交系群体,是由重组近交系组成的分离群体[8 ],通常从F2代开始采用单粒传的方法来建立。由于自交的作用是使基因型纯合化,RIL群体中每个株系都是纯合的,因而RIL群体是一种可以长期使用的永久性分离群体。目前RIL群体被广泛应用,能保持单个纯合基因型个体。
1.2.3 近等基因系(Near isogenic line,NIL) 群体将多个QTL位点分解成单个孟德尔遗传因子,将数量性状转化为质量形状,从而可以对主效QTL进行精细定位和图位克隆[8 ]。
1.2.4 深度杂交系(Advanced intercross lines,AIL)
群体AIL开始于F2群体,F1杂交后裔继续杂交一定数目的世代,F2 ∶ 3继续杂交后代(与RIL近似,但是远交,而不是近交)[8 ]。
1.2.5 双单倍体(Doubled haploid,DH) 群体指高等植物的单倍体经过染色体加倍形成的二倍体。DH群体产生的途径很多,亦因物种不同而异,最常见的方法是通过花药培养,即取F1 植株的花药进行离体培养诱导产生单倍体植株,然后对染色体进行加倍产生DH植株。也可通过单倍体诱导系诱导,通过自然加倍或化学药剂处理加倍产生DH群体[8 ]。
1.2.6 回交(Back Cross,BC)群体 也是常用的作图群体,如BC1为A与B的杂交F1代与A或者B回交产生的子代,直接反映了F1代配子的分离比例[8 ]。
1.2.7 F2群体 为A与B的杂交F1代经自交产生的子代,是一种暂时性分离群体。特点是群体内个体间基因型不同,各个体的基因型杂合。这类群体的优点是不但可提供丰富的遗传信息,而且可以用来估算加性效应及显性效应。F2群体或衍生的F2 :3群体是最常用的作图群体[8 ]。
1.2.8 连锁群体(Linkage group) 标记组成的序列,染色体是碱基组成的序列,连锁群上的标记只是存在多态性的碱基位点,即SNP,所以来自于同一条染色体的标记一定构建到同一连锁群上[8 ]。
2 基于遗传群体进行基因定位方法
目前,定位质量性状控制基因主要利用近等基因系分析法、连锁分析法和混合分组分析法等途径,在不同作物基因定位及遗传改良中均有成功案例。
2.1 近等基因系分析法(Near isogenic line analysis)
将遗传背景相近或相同、多个农艺性状相似、个别染色体位置存在差异的材料称之为近等基因系。其主要获得手段是将两个具有不同目标表型的亲本P1和P2杂交,再与P1或P2多代回交后筛选得到在目标表型上有差异的品系,这样,品系以及品系间与轮回亲本间就构成了近等基因系。在育种中,亲本轮回改良或品种特性改良应用最多的就是近等基因系分析法[12 - 13 ],当市场推广品种缺少某个优良性状时(如不抗虫、易感病),常采用轮回转育方法将外源品种中含有目标性状的抗性基因导入到现有品种中,提高该品种的综合抗性;若是杂交种,只改变存在缺陷性状的父本或母本之一,以保证杂交种其他农艺性状的稳定性。因此将用于多次回交的亲本称为受体亲本或轮回亲本,是目标表型的接受者,仅用于首次杂交时的亲本称为供体亲本或非轮回亲本,是目标表型的提供者。如此,多代回交的结果就是不断提高后代中轮回亲本的遗传基因,减少供体材料的遗传成分,逐渐向轮回亲本遗传背景纯合,7~8代后理论上除了含有需改良的目标性状基因片段外,其他染色体遗传信息与轮回亲本几乎相同(育种上讲就是拥有了99%以上的轮回亲本血缘)。为了加快回交后代基因组恢复成轮回亲本的速度,在每代选择继续回交的植株时,除了要保证含有供体目标基因外,应尽量选择形态上与轮回亲本接近的植株。因此,改良的品系与轮回亲本间实际上构成了一对近等基因系,通过持续的自交保持目标性状位点的杂合可以建立成对的近等基因系,利用近等基因系分析法可以克隆控制重要农艺性状的候选基因。
2.2 连锁分析法(Linkage analysis)
用连锁分析法定位QTL的基本原理是经典遗传学中的连锁分析(两点测交、三点测交),利用与QTL连锁的分子标记进行推断[14 - 15 ]。因为当一个分子标记与QTL连锁时,倾向于一起遗传,这样就可以借助分子标记信息近似估计QTL在群体中的传递情况。还可利用区间定位方法。与单标记分析法比较,区间定位法能大大提高QTL的检测效率,并能较准确的估计出QTL的位置和效应值。区间定位已成为目前QTL检测中的标准方法。QTL连锁分析常用的统计分析方法有区间作图法(IM)、复合区间作图法(CIM)、混合线性模型(MLM)、完備区间作图法(ICIM)等。关联分析需要依赖作图群体,构建遗传图谱周期长,成本较高。
2.3 关联分析法(Correlation analysis)
关联分析可以解决连锁分析在挖掘种质资源等位变异时效率低、成本高、在群体数量和研究目标性状方面依赖作图群体等缺陷。GWAS分析是对多个个体全基因组范围的遗传变异(标记)进行检测,获得基因型,进而将基因型与可观测的性状即表型进行群体水平的统计学分析,根据统计量或显著性P值筛选出最有可能影响该性状的遗传标记,定位与性状相关的遗传位点或挖掘与性状变异相关的候选基因,在群体水平上解析性状遗传基础[10, 16 ]。关联分析具有许多优点,如可以直接使用自然群体或种质资源,不需要专门构建作图群体;检测效率高,可同时检测同一座位的多个等位基因;节省研究时间且分辨率高。但相应地,GWAS 也存在一定的缺点,如当群体结构分化明显时容易造成假阳性,需要足够多的样本才能保证P值足够低,来避免假阴性和假阳性。同时,精确的表型鉴定是GWAS成功定位的重要保障。
2.4 离体分组混合分析法(Bulked segregate analysis, BSA)
离体分组混合分析法是1991年由R. W. MICHELMORE[10 ]在莴苣上首次应用的一种快速定位控制目标性状基因的方法。取F2群体中具有极端表型的15~100个单株等量混合其DNA形成2个DNA池,然后在亲本和两个池之间进行标记多态筛选。如果某个标记在亲本和混池之间具有一致的多态,则该标记很可能和性状连锁,通过F2群体对这些筛选到的多态标记进行基因型分析即可完成对目标基因的定位,而不需要对每个标记在群体里进行基因型分析。BSA的方法常用于单个QTL、突变位点定位等,BSA建立的混池因为经过了目标性状筛选,所以保证了2个混池之间除了目标性状外其他遗传背景基本相同,仅目标性状关联的DNA区段存在差异,所以两个混池又被称为近等基因池。该方法主要用于植物性状的初定位,适用于单基因的质量性状和主效基因的数量性状上,常用分离群体为F2、BC、RIL、DH群体等,常用的分子标记有RAPD、RFLP、AFLP、SSR、SNP、InDel等。
BSA技术应用广泛的主要有以下4种方法,分别是MutMap[17 - 18 ]、MutMap+[19 ]、MutMap-Gap[20 ]、QTL-seq[21 - 22 ],他们的基本原理相同,即针对研究的目标性状,选择表型差异显著的亲本构建出分离群体(或家系群体),再从分离群体中选取目标性状表型极端的一定数量的单体,混合构建2个DNA池(DNA pools)。通常以双亲的DNA作为对照(参考基因组以利于对实验结果进行正确的分析和判断),比较2个DNA池之间的差异,2个DNA池之间的差异片段即为候选区域,所关注的基因或者QTL可能存在于该候选区域中[23 ]。
3 不同MutMap方法及应用
3.1 MutMap方法
3.1.1 MutMap定义及适宜条件 MutMap是基于高通量第二代测序技术的全基因组测序(Whole genome sequencing,WGS)发展起来的新的正向遗传学基因定位和遗传分析方法[17 ]。2012年Abe 等[17 ]开发的MutMap方法应用于水稻性状筛选改良,利用甲基磺酸乙酯(ethylmethanesulfonate,EMS)诱变处理野生稻后获得稳定遗传突变体,且目标性状由隐性单基因控制。取突变表型个体DNA等量混合获得突变体DNA池(选择30~200株不等)进行全基因组重测序,混池测序数据比对诱变亲本基因组获取SNP位点,计算SNP指数,指数峰作为目的性狀关联的目的基因所在的候选位置。利用该技术成功定位到4个水稻半矮秆、1个雄性不育、2个叶色淡绿性状关联基因在染色体上的遗传位置[17 ]。
3.1.2 MutMap原理及分析 利用二代测序技术,根据BSA极端性状混池测序原理[17,23 ],分别抽提具有极端表型性状植株的亲本及子代植株DNA或RNA建库测序。在分离群体搭建过程中,子代会根据表型进行选择,筛选出突变型子代池和野生型子代池。根据遗传连锁交换定律,子代池的基因型会和表型产生共分离,反应在物理图谱层面,与表型连锁的染色体区段会和不连锁的染色体区间产生稳定的SNP-指数差异(SNP-index)。SNP指数的概念:SNP-index =突变SNP的reads数/对应SNP位点的总reads数。差异大小范围为0~1。如果SNP-index=0,说明该池中仅包含来自参考基因序列的亲本基因组片段;如果SNP-index=1,说明该池中仅仅包含来自于另一个亲本(突变体)的基因组片段;如果SNP-index=0.5,说明该池中存在2个亲本的基因组片段,此子代混池中SNP来自2个亲本的基因组的频率一致(图1)。操作及分析步骤:①筛选亲本间纯合差异的SNP位点;②子代SNP根据亲本的基因型,确定突变亲本来源的Reads,计算SNP-index;③只有突变子代池(Highbulk)的群体采用SNP-index定位,同时存在突变子代池和野生子代池(Lowbulk)的群体,采用ΔSNP-index定位,即将突变池的SNP指数减去野生池的SNP指数以获得每个位点的SNP指数差值,即Δ(SNP-index);④定位时以每15个点为窗口,5个点步移统计该窗口内所有SNP-index平均值,做出红色拟合线,排除部分噪音影响,使连锁区域峰值更加明显;⑤ΔSNP-index的群体可以利用二项分布检验Highbulk和Lowbulk的覆盖深度,考虑其混池数量以及群体类型,算出其统计学上95%的和99%的置信线;⑥ΔSNP-index定位时,拟合线超出置信线的染色体区域为可能的表型连锁区域。根据拟合线超出染色体置信区间,判断候选基因位置[17 ]。测序完成后,要对测序结果进行系统分析,常用的且最重要的分析参数有以下6种:一是SNP,单核苷酸多态性(Single nucleotide polymorphism),主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性;二是InDel,插入/缺失(Insertion/Deletion)基因组中小片段的插入和缺失序列;三是SV,基因组结构变异(Structure variation),染色体结构变异是指在染色体上发生了大片段的插入和缺失、重复复制、翻转颠换、易位等;四是CNV,基因组拷贝数变异(Copy number variation),是基因组变异的一种形式,通常使基因组中大片段的DNA形成非正常的拷贝数量;五是基因组测序深度,测序得到的总碱基数与待测基因组大小的比值;六是基因组覆盖率,由于大片段拼接的gap、测序读长有限、重复序列等问题的存在,测序分析后组装得到的基因组序列通常无法完全覆盖所有区域,覆盖度就是最终得到的结果占整个基因组的比例。
3.1.3 MutMap应用实例 2011年日本地震导致海啸爆发、海水倒灌农田,使当地原本肥沃的土地变成了盐碱地,水稻产量大幅下降。科学家采用EMS诱变的方法对当地水稻品种“Hitomebore” 进行了处理,F1代自交得到的 F2群体,获得了耐盐型单基因控制耐盐突变株,野生型和突变型表型个体数的比值约为 3∶1,符合孟德尔隐性单基因控制遗传定律。选取带有目标性状的子代个体构建混合池并测序,将测序结果与野生亲本基因组比对,得到SNP位点,通过Δ(SNP_index)的方法将目标性状定位在6号染色体2.76~8.57 Mb之间。该候选区域其中1个SNP的突变使一个无义色氨酸密码子突变为终止子,在筛选到与耐盐性状相关的基因OsRR22,导致突变体hst1产生耐盐表型。该研究缩短了耐盐品种水稻的育种进程,将对当地水稻产量恢复做出巨大贡献[18 ]。
3.2 MutMap+方法
3.2.1 MutMap+适宜条件及原理 常规的MutMap方法是将 M3~M5世代的突变体与野生型亲本株系进行杂交构建后代群体,在此基础上进行BSA分析。对于人工杂交困难的突变体,以及发育早期死亡或不育的突变体,因为无法直接构建子代群体,MutMap方法就不适用了。为了解决这个问题,Fekih等[19 ]开发了MutMap+方法:隐性纯合M1代株系不育或死亡,因此利用杂合的M1代自交产生的杂合后代来保存变异基因,并在构建群体时重点关注那些自交后代表型发生分离的株系。在导致表型的突变位点上野生型株系理论上有2/3是杂合的。以水稻为例,简单阐述MutMap+方法的原理(图2)。
3.2.2 MutMap+应用实例 应用该方法不杂交只自交,在杂合的株系自交获得的后代群体中,野生型表型和隐性纯合突变表型分离比例约为3∶1。将野生型 M1后代个体自交,收集杂合的M1株系自交获得的M2代群体。同样收集杂合的M2代自交获得的M3群体。根据表型将M3株系分为两组,构建2组混合DNA池,并进行全基因组重测序。混池测序数据比对野生型亲本基因组获取SNP位点,2个混池分别计算SNP-index,Δ(SNP-index)峰作为目的性状关联的目的基因所在的候选位置。该方法主要用来定位早衰、致死、不育、白化等性状的定位分析,也用来分析花蕊太小或去雄难度大而尚未建立有效的人工杂交物种。例如谷子:利用MutMap+方法,Fekin等找到了表型为早衰和过早死亡的突变体Hit9188的候选基因OsNAP6,并且找到了表型为苗白化和过早死亡的突變体Hit11440 的候选基因Os08g0139100[19 ]。
3.3 Mutmap-Gap方法
3.3.1 Mutmap-Gap适宜条件 Mutmap或Mutmap+都需要根据参考基因组序列先构建一个诱变亲本的Pseudo-genome。如果野生型亲本的基因组和参考基因组的序列有结构性差异,而导致性状改变的区域位点恰巧不在参考基因组序列上,相对参考基因组是一个大的插入片段,是参考基因组上的缺乏的区域(Gap),直接做Mutmap分析将得不到关联位点的全部信息。为了鉴别在参考基因组序列Gap内的变异,Takagi等[20 ]开发了Map-Gap方法。该方法将MutMap方法与对基因组gap区域的从头(de novo)组装相结合,对这类特异的突变基因进行定位和分离。MutMap-Gap将MutMap方法得出的靶基因组区域的gap通过de novo组装补齐,以判断不在参考基因组上变异候选区段位置(图3)。
3.3.2 Mutmap-Gap原理及应用 首先将比对到候选区域的临近区域和未对应参考序列的reads序列合并起来做de novo组装,将组装得到scaffold序列和pseudo-genome序列一起当作参考序列(图3),重新比对计算SNP指数,关联定位区域。如突变位于P特异性基因组区域内,仅通过MutMap分析无法识别。因为定位区域相对参考基因组是一个新的发现,参考序列里面没有基因相关的信息,通过对P的全基因组测序及与参考基因组的重新组装pseudo-genome序列,需要对该区域进行基因预测和注释,以得到目的基因。一般情况下Mutmap或Mutmap+没定位到结果,可以尝试用Mutmap-Gap。
3.4 QTL-Seq方法
3.4.1 QTL-Seq方法原理及操作 MutMap系列仅适用于质量性状,对于数量性状,产生了QTL-seq法。QTL-seq是一种在极端表型混池高通量测序的基础上,根据极端表型池间SNP频率的差异进行QTL/基因定位的方法,是一种结合BSA与NGS 来快速定位单个数量性状的方法[21 - 22 ]。具体操作:选取目标性状差异大的两个亲本杂交,产生分离群体如F2、RILs、DH 等,目标性状理论上将会呈现正态分布选取目标性状表型极端的20%的比例的个体分别混合成2个表型极端池,进行重测序。以对照亲本的基因型为参照,计算子代极端池中的SNP-index[17 ]。若该参数为0,代表子代所有测到的Reads都来自野生亲本;若该参数为1,代表子代所有Reads都来自突变亲本;该参数为0.5,说明该池中存在2个亲本的基因组片段。一般而言,大部分位点的SNP index值在0.5左右的区域,但如果某SNP与目标QTL连锁,导致SNP-index的值偏离0.5。比较ΔSNP-index,候选区间内的ΔSNP-index应该接近于1,从而将QTL定位到了基因组上的某一区域[18 - 19 ](图4)。
3.4.2 QTL-Seq方法应用实例 QTL-seq定位黄瓜早花农艺性状:采用BSA混样策略对10株F2子代群体极端性状(早花和晚花)的样品混合的DNA池,及其亲本进行基因组重测序。通过全基因组扫描SNP,分析频率差异,检测F2群体早花性状的QTL,找到了一个位于早花QTL Ef1.1中的候选基因。利用F2群体,SSR标记构建的遗传图谱进行QTL定位,两种策略结合,将Ef1.1缩小到980 Kb,其中包含基因Csa1G651710,与FT基因同源[22 ]。
3.5 4种不同MutMap方法的比较
从表1可以看出,MutMap、MutMap+、Mut Map-Gap、QTL-seq这4种方法在样本材料、亲本类型、杂交方式、所需测序样本、适用范围等不尽相同,但均可快速获得与目标基因连锁的分子标记,且遗传定位的周期短、效率高。基于诱变突变体的MutMap方法、自交的MutMap+方法、用于识别基因组缺失区间变异的MutMap-Gap方法以及用于定位数量性状基因座的QTL-seq方法等均不需要建立繁琐的后代定位群体,加快了对重要表型变异位点的识别过程,缩短了育种时间,大大加快了新品种的开发速度。
4 小结与展望
由于MutMap方法的应用使得基因得以快速定位,這些基因资源将有可能促进玉米、水稻等作物的遗传改良[1 - 2 ]。基于WGS的MutMap方法的正向遗传学研究,不仅有利于QTL/基因的定位,也利于新的基因资源在作物育种中的应用。早在1991年,Michelmore等[10 ]就建立了BSA法,克服了许多作物没有或难以创建相应的NIL群体的限制,在自交和异交作物中均有广泛的应用前景。对于尚无连锁图或连锁图饱和程度较低的植物,BSA法也是快速获得与目标基因连锁的分子标记的有效方法,该方法的优点是遗传定位的周期短且效率高。在此基础上扩展的新方法也不断出现,如基于自交的MutMap+、用于识别基因组缺失区间变异的MutMap-Gap,以及用于定位数量性状基因座的QTL-seq方法等。这些方法的优点是不需要建立繁琐的后代定位群体,甚至不依赖于遗传杂交和任何连锁信息,加快了对重要表型变异位点的识别过程。Mutmap采用EMS诱变技术将育种时间缩短至传统育种的1/5左右,大大加快了新品种的开发,通过利用MutMap对突变体进行遗传定位研究,可以快速鉴别出靶基因[23 - 24 ]。
过去,人们依靠传统育种方法获取作物新品种,根据选择的特性确定植物亲本,然后通过杂交、回交或者直系筛选程序来完成。传统育种方法存在着极大的盲目性、经验性、不确定因素和长周期等问题。为了实现育种4.0的目标,需要为分子育种制定一个育种路线图。应用新的生物技术,依托人工智能、基因组测序、基因编辑等相关技术,实现玉米组学基因型与表型大数据的快速积累。通过遗传变异等数据的整合,实现作物性状调控基因的快速挖掘与表型的精准预测。通过人工改造基因元器件与人工合成基因回路,使作物具备新的抗逆、高效等生物学性状,创建智能组合优良等位基因的自然变异、人工变异、数量性状位点的育种设计方案,最终实现智能、高效、定向培育新品种[25 ]。分子育种的精准实施可帮助育种家在最短的时间里实现以下目标。①大大缩短育种时间,加速育种进程。比如玉米自交系选育中,通过常规育种回交6代及以上才能达到99%纯合的后代,而分子标记选育只需要3代就可以达到,利用单倍体诱导技术只需要2代就能达到100%纯合。②最大限度地绕过物种生殖隔离的障碍,实现生物界遗传物质的自由交流。利用基因重组操作技术,将不同物种中特定的基因与载体结合,可以导入到其他物种中表达,发挥其基因功能,利用转基因技术让现存的作物品种快速适应新环境[26 ]。如抗棉铃虫转基因棉花的普及和大面积种植推广,以及抗旱耐盐碱、抗除草剂等玉米品种的获得,都大大降低了人工打药、锄草、灌溉等成本,提高了产量和收入,且转基因观赏园艺植物在休闲观光农业中被广泛应用。③可帮助育种从多个目标性状带有经验性的多代重组选择,转变成针对目标性状进行基因编辑的精准性状选择和改良,可将主效基因及其优异等位基因集于一个骨干自交系或品种中,大大提高了育种效率。如孟山都公司利用分子标记将多个不同的抗旱位点聚合育成高抗旱玉米品种(gene stacking),是复杂性状通过分子标记获得育成品种的成功案例[3 ]。刘忠祥[27 ]系统阐述玉米株高主效QTL定位研究进展及与株高相关基因的功能与响应途径, 对玉米分子育种的选育工作有积极借鉴作用。④随着人们对身体健康,美容保健等方面的重视,育种家通过分子育种可选育功能保健型(低脂低糖、高蛋白、高氨基酸等)品种,以满足人们通过饮食对特殊营养成分需求,如“黄金大米”,其β-胡萝卜素的含量是普通大米的23倍。⑤基因组编辑技术作为一项变革性的新兴技术,尤其是CRISPR/Cas 9基因编辑技术的兴起和广泛应用,将推动新技术产品产业化的发展[28 - 29 ]。中国科学院高彩霞团队等联合攻关,阐明了小麦新型mlo突变体既抗白粉病又高产的分子机制,并通过CRISPR/Cas9多重基因组编辑,使主栽小麦品种快速获得广谱抗白粉病的优异性状[30 ]。然而,没有前期的对关键基因进行克隆和功能的深入剖析,根本谈不上DNA水平的分子标记辅助选择育种。因此,加强农业种质资源保护和开发利用,创新农作物分子改良与技术应用体系,加强作物分子育种方面的应用基础性研究是未来提高中国种业发展水平最为重要的步骤和方向[31 - 35 ]。
参考文献:
[1] 种 康,李家洋. 科学发展催生新一轮育种技术革命[J]. 中国科学:生命科学,2021,51(10):1353-1355.
[2] 景海春,田志喜,种 康,等. 分子设计育种的科技问题及其展望概论[J]. 中国科学:生命科学,2021,
51(10):1356-1365.
[3] 薛勇彪,种 康,韩 斌,等. 创新分子育种科技支撑我国种业发展[J]. 中国科学院院刊,2018,33(9):893-899.
[4] PINGALI P L. Green revolution:impacts,limits,and the path ahead[J]. Proceedings of the National Academy of Sciences of the United States of America,USA,2012,109:12302-12308.
[5] 周文期,连晓荣,刘忠祥,等. 玉米株高和穗位高的调控机理研究[J]. 分子植物育种,2021,19(23):7965-7976.
[6] 周文期,连晓荣,周玉乾,等. EMS诱变玉米自交系种质创新应用[J]. 玉米科学,2020,28(6):31-38.
[7] 周文期,王晓娟,寇思荣,等. 玉米叶夹角形成的分子调控机理研究[J]. 土壤与作物,2019,8(3):339-348.
[8] 朱玉贤,李 毅,郑晓峰,等. 现代分子生物学[M]. 5版. 北京:高等教育出版社,2019.
[9] COULSON A,SULSTON J,BRENNER S,KARN J. Toward a physical map of the genome of the nematode Caenorhabditis elegans[J]. Proceedings of the National Academy of Sciences of the United States of America,USA,1986,83:7821-7825.
[10] MICHELMORE R W,PARAN I,KESSELI R V. Identification of markers linked to disease-resistance genes by bulked segregant analysis:a rapid method to detect markers in specific genomic regions by using segregating populations[J]. Proceedings of the National Academy of Sciences, 1991,88(21):9828-9832.
[11] VISSCHER P M,WRAY N R,ZHANG Q,et al. 10 years of GWAS discovery:Biology,function,and translation[J]. American Journal of Human Genetics, 2017,101(1):5-22.
[12] 张 叶,王梓钰,邢跃先,等. 利用近等基因系定位玉米无叶舌基因的研究[J]. 玉米科学,2021,29(3):29-35
[13] 傅大雄,阮仁武,刘大军,等. 近等基因系法对小麦显性矮源的研究[J]. 中国农业科学,2007,40(4):655-664
[14] 王 芸. 关联分析和连锁分析定位控制水稻有效穗数QTL [D]. 北京:中國农业科学院,2018.
[15] 田 玉,马春红,宋丽华,等. 数量遗传性状基因定位方法研究进展[J]. 河北农业科学,2021,25(5):88-91.
[16] 涂雨辰,田 云,卢向阳. 全基因组关联分析在植物中的应用[J]. 化学与生物工程,2013,30(6):1672-5425.
[17] ABE A,KOSUGI S,YOSHIDA K,et al. Genome sequencing reveals agronomically important loci in rice using MutMap[J]. Nature Biotechnology,2012,30(2):174-178.
[18]TAKAGI,H.,TAMIRU,M.,ABE,A. et al. MutMap accelerates breeding of a salt-tolerant rice cultivar[J]. Nature Biotechnology,2015,33,445-449.
[19] FEKIH R,TAKAGI H,TAMIRU M,et al. MutMap+:Genetic mapping and mutant identification without crossing in rice[J]. PLoS One,2013,8(7):e68529.
[20] TAKAGI H,UEMURA A,YAEGASHI H,et al.,MutMap-Gap:whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii[J]. New Phytologist,2013,200(1):276-283.
[21] TAKAGI H,ABE A,YOSHIDA K,et al. QTL-seq:rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations[J]. Plant Journal,2013,74(1):174-183.
[22] LU H,LIN T,KLEIN J,et al. QTL-seq identifies an early flowering QTL located near Flowering Locus T. in cucumber[J]. Theoretical and Applied Genetics,2014,
127:1491-1499
[23] 袁金红,李俊华,袁娇娇,等. 基于全基因组测序的MutMap方法在正向遗传学研究中的应用[J]. 遗传, 2017,39(12):1168-1177
[24] 陆才瑞,邹长松,宋国立. 高通量测序技术结合正向遗传学手段在基因定位研究中的应用[J]. 遗传,2015,37(8):765-776.
[25] 王向峰,才 卓. 中国种业科技创新的智能时代—“玉米育种4.0”[J]. 玉米科学 2019,27(1):1-9.
[26] 王红梅,陈玉梁,石有太,等. 中国作物分子育种现状与展望[J]. 分子植物育种,2020,18(2):507-513.
[27] 刘忠祥. 玉米株高主效QTL定位研究综述[J]. 甘肃农业科技,2018(9):62-69.
[28] 卢俊南,褚 鑫,潘燕平,等. 基因编辑技术:进展与挑战[J]. 中国科学院院刊,2018,33(11):1184-1192.
[29] 陈云伟,陶 诚,周海晨,等. 基因编辑技术研究进展与挑战[J]. 世界科技研究与发展,2021,43(1):8-23.
[30] LI S N,LIU D X,ZHANG Y W,et al. Genome-edited powdery mildew resistance in wheat without growth penalties[J]. Nature,2022,602:455-460.
[31] 郑怀国,赵静娟,秦晓婧,等. 全球作物種业发展概况及对我国种业发展的战略思考[J]. 中国工程科学,2021,23(4):45-55.
[32] 周喜旺,刘鸿燕,王 娜,等. DNA分子标记技术在小麦遗传育种中的应用综述[J]. 甘肃农业科技,2017(5):64-68.
[33] 李闻娟,齐燕妮,王利民,等. 运用SRAP分子标记对胡麻杂交种纯度的鉴定研究[J]. 甘肃农业科技,2019(9):59-62.
[34] 陈 琛,罗俊杰,陈卫国. 利用SSR技术快速鉴定2个辣椒杂交品种纯度[J]. 甘肃农业科技,2020(4):53-58.
[35] 刘新星,欧巧明,罗俊杰,等. 利用SSR标记鉴定当归的真实性[J]. 甘肃农业科技,2020(7):43-48.
收稿日期:2022 - 03 - 09
基金项目:国家自然科学基金(32160490、31860384);2020年甘肃省科协青年科技人才托举工程项目;甘肃省农业科学院创新专项-博士基金(2020GAAS34)。
作者简介:周文期(1985 — ),男,甘肃静宁人,副研究员,博士,主要从事玉米遗传育种及基因功能研究工作。Email: zhouwenqi850202@163.com。
通信作者:周玉乾(1979 — ),男,甘肃靖远人,研究员,主要从事玉米育种工作。Email:yuqianzhou2008@163.com。
