不同类型儿童吉兰-巴雷综合征变异型诊治报道
2022-05-07何运元王院方郭爱萍
何运元,王院方,郭爱萍
(濮阳市安阳地区医院儿科,河南 濮阳 455000)
吉兰-巴雷综合征(GBS)是最常见引起儿童急性弛缓性麻痹的自身免疫性疾病,主要表现为急性进展、对称性肌无力及腱反射减弱或消失,呈单相病程[1]。但随着近几十年来对GBS的研究,这一经典诊断标准受到极大挑战。GBS变异型临床表现不典型,早期多无肌力下降,临床症状复杂、多变,涉及多个系统,临床上常被误诊及延误治疗。Miller-Fisher综合征(MFS)、急性泛自主神经病及急性感觉神经病是GBS的特殊亚型,其发病率低,误诊率高,国内仅有少量报道[2-4]。本文通过对不同类型儿童GBS变异型的临床资料进行分析并总结治疗经验,以期对临床工作提供帮助。
1 资料与方法
1.1资料
1.1.1一般资料 本院2016年5月至2020年5月共收治MFS 5例,急性泛自主神经病3例,急性感觉神经病4例,回顾性分析临床资料。所有患者知情同意,符合《赫尔辛基宣言》的基本原则。
1.1.2纳入标准 (1)儿童急性起病,快速进展,达运动障碍高峰小于4周;(2)符合MFS、急性泛自主神经病及急性感觉神经病临床特点,按照中华医学会神经病学分会制定标准[5];(3)排除脊髓灰质炎;(4)肌张力正常或者轻度减退;(5)脑脊液蛋白-细胞分离现象;(6)自限性;(7)电生理检查可正常或感觉神经异常;(8)排除其他病因。
1.2方法
1.2.1观察指标 分析患者的一般资料、起病情况、临床特点、病情演变(最大运动障碍高峰时间、应用呼吸机情况)、脑脊液检查、神经电生理,分析其治疗措施及预后。
1.2.2功能障碍评分 采用Hughes残疾等级改变判断患儿最大运动障碍[6]。0分:正常;l分:不影响正常活动;2分:无扶助可独自行走大于5 m;3分:扶持或借助工具行走小于5 m;4分:床上或轮椅上活动;5分:需要呼吸机支持;6分:死亡。
2 结 果
2.1一般资料情况 12例GBS变异型患儿中男7例,女5例,男女比例为1.4∶1.0;3~<8 岁10 例,8~10岁2例。多在学龄前期发病,起病至确诊时间4~60 d,中位时间为19 d。
2.2临床特点 12例GBS变异型患儿中83.3%(10例)存在感染诱因,其中上呼吸道感染4例,胃肠道感染3例,疱疹性咽峡炎2例,支原体肺炎治疗病史1例;病毒感染8例,占66.7%。首发症状具体如下:(1)MFS,眼球运动异常2例,眼睑下垂伴步态不稳1例,视物重影伴共济失调2例;(2)急性泛自主神经病,头晕伴尿潴留1例,行走不稳伴视物模糊1例,抽搐后意识障碍1例;(3)急性感觉神经病,肢体疼痛2例,肢体麻木2例。所有患儿早期均无肌力下降,查体均存在腱反射减弱或消失。2周内出现最大运动障碍为85%,其中1周内达峰为40%,少数病例在起病后30 h左右即达高峰,1例急性泛自主神经病给予呼吸机支持,机械通气时间为50 h。至确诊时肌力正常5例,肌力Ⅲ~Ⅳ级5例,肌力Ⅱ级2例。见表1。
2.3辅助检查 (1)脑脊液出现典型蛋白-细胞分离80%(10例)出现在第2周,第1周即出现的占20%(2例,其中均为急性感觉神经病);脑脊液常规:潘氏球蛋白定性实验均阳性,白细胞计数(0~5)×106L-1;脑脊液生化:脑脊液蛋白 0.787~1.612 g/L[平均(1.1±0.28)g/L]。(2)血、脑脊液神经节苷脂抗体送检6例,其中1例急性泛自主神经病血清抗GM1抗体IgM(+),其余均为阴性。(3)肌电图:肌肉电传导减慢(左右尺神经CMAP波幅稍低)1例,轴索损伤为主的周围神经病变3例,感觉神经动作电位波幅下降(考虑脱髓鞘改变)5例,正常3例。(4)头颅及脊髓影像学检查:头颅影像学检查阴性10例,顶叶皮层异常信号及大枕大池1例(已进行血氨、乳酸、遗传代谢、感染等相关检查、筛查,排除颅内感染、占位、代谢性疾病);行脊髓MRI检查:腰椎段脊髓受累,腰段神经根明显强化2例,脊髓轻度水肿(胸椎段)3例,其余均阴性。
2.3功能障碍评分结果 MFS 5例中Hughes功能评分分级小于3分4例,1例4分;急性泛自主神经病3例中1例5分,1例3分,1例2分;急性感觉神经病4例中3例2分,1例3分,1例4分;统计小于或等于3分占75%,>3分占25%。见表1。
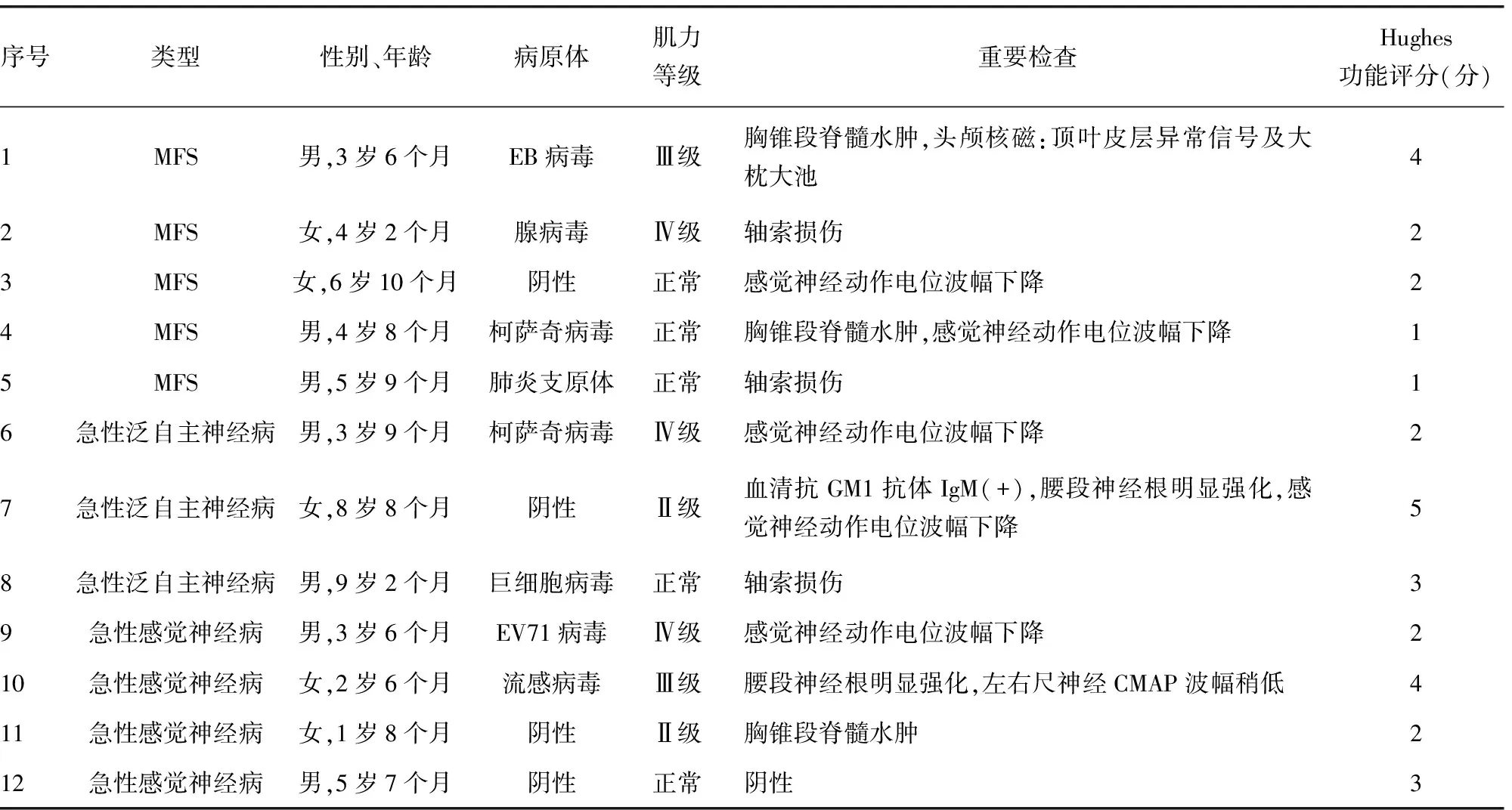
表1 12例GBS变异型临床特点、辅助检查及Hughes功能评分比较
2.4治疗及转归 6例给予1个疗程免疫球蛋白(总量2 g/kg,分3 d输注)治疗,4例应用2个疗程免疫球蛋白治疗(剂量2 g/kg,间隔7~14 d),2例伴有明显神经根疼痛,采用免疫球蛋白联合甲泼尼龙20 mg/kg,每天1次静脉滴注×3 d(最大量小于500 mg/d),随后减量为甲强龙(甲泼尼龙琥珀酸钠)10 mg/kg,每天1次静脉滴注×2 d,后改为泼尼松片[2 mg/(kg·d)]口服,每3天减量5~10 mg,直至减停。4例患儿在1周肌力开始恢复,3例在2周内肌力逐渐恢复,本组无1例死亡,出院前5例(表1中序号1、7、8、10及11)进入康复训练阶段,随访6个月,4例恢复正常,1例(序号10)因家庭经济及其他原因,遗留共济失调、头晕不适。
序号7,Hughes分级5分,最初以胃肠道感染症状(发热伴呕吐)起病,呼吸麻痹,给予气管插管、机械通气50 h,误诊“病毒性脑炎”,起病第3天予丙种球蛋白免疫(总量2 g/kg,分3 d输注)治疗1个疗程,病程中第4、9天均行腰椎穿刺脑脊液检查均阴性,头颅核磁阴性。治疗1周后意识恢复,肌力下降逐渐显现,起初左上肢肌力Ⅱ级,其余肢体肌力Ⅲ级,后肌力恢复,出现头晕、少汗、直立性低血压等自主神经紊乱症状,病程第2周复查脑脊液常规:潘氏球蛋白定性实验阳性,白细胞3×106L-1;脑脊液生化:脑脊液蛋白1 028 mg/L;典型蛋白-细胞分离现象,血清神经节苷脂抗体:抗GM1抗体IgM(+),肌电图示周围神经受损。腰椎磁共振成像(MRI)平扫+增强提示腰段脊髓受累,腰段神经根明显强化。最终确诊患者急性泛自主神经病。间隔7 d后再次给予第2个疗程免疫球蛋白应用,总治疗时间4周,治愈出院。
3 讨 论
随着GBS亚型的不断更新及报道,不典型表现逐渐被人们认知。MFS、急性感觉神经病和急性泛自主神经病作为其中少见的类型,其首发症状多无肌力下降,其独有的复杂临床特点,几十年内未被收录到GBS诊断系统内[7]。随着研究的深入,2019年中华医学会神经病学分会对GBS亚型进行了更新,这3类变异型[5]被归纳入GBS谱系中,作为GBS变异型,对其临床特点等进行总结。
对于变异型GBS的诊断,详细的病史及神经系统查体至关重要,感染诱因、感觉障碍、脑脊液检查、相关周围神经病抗体(如抗神经抗原抗体检测、抗神经节苷脂抗体等)的出现及神经电生理依据则是GBS诊断的支持证据。本研究患儿男性较多,年龄多在学龄前期,83.3%患儿起病前有感染诱因,多数存在呼吸道感染,而其中EB病毒、腺病毒等病毒检出率高(占66.7%)。以往研究认为,GBS是感染后免疫介导的神经损伤,常见病原菌为空肠弯曲杆菌[8],其次为巨细胞病毒、EB病毒、肺炎支原体等,在儿童中尚见少量报道与之相一致,且病毒感染较成人发挥重要作用[4,9-10],本研究与之相符。本研究中3类GBS变异型首发症状以眼外肌麻痹、共济失调、自主神经及感觉神经受累为特征性表现,早期均无肌力下降,至确诊前肌力Ⅱ级仅占16.7%,85%的患儿在2周内达最大运动障碍,其中最短达峰时间为30 h(Hughes功能评分5分),急速进展,给临床诊断带来巨大困难,本研究确诊周期最长2个月;神经电生理异常占75%,以感觉神经运动电位异常(脱髓鞘改变)多见。因本研究样本量小、有的疾病在确诊早期即给予了免疫治疗等因素,周围神经病相关抗体检出率低,仅1例检出抗GM1抗体IgM(+),仍需大样本进一步验证。
变异型GBS治疗同经典型一致,主要为免疫治疗,静脉滴注丙种球蛋白及血浆置换是同等有效的[11],由于丙种球蛋白治疗并发症更少,临床上较常用。对于Hughes功能评分小于或等于2分的轻症GBS,有研究显示免疫治疗与支持治疗相比,远期预后无显著差异,但也有研究显示早期丙球应用可明显改善运动障碍及缩短住院时间[12],但考虑其病情可能进一步进展,仍需积极免疫治疗,改善预后;对于Hughes功能评分大于或等于3分的GBS患者,2周内给予丙种球蛋白治疗,可收获最大疗效,考虑与丙种球蛋白可结合致病性的抗神经节苷脂抗体,阻断免疫活化有关系[11]。本组12例GBS患儿均在2周内给予免疫球蛋白治疗,7例肌力下降患儿均在用药后2周有不同程度恢复,短期预后(6个月)良好;研究显示,约有10%的患者在给予丙种球蛋白或血浆置换治疗获得短暂缓解或病情趋于稳定后,会再次加重,称为治疗相关波动(TRF),由于这部分患者的自身免疫反应延长,从而导致更长久的神经损伤,因此治疗时间也需要延长。WALGAARD 等[13]发现,出现TRF的GBS预后疗效受益于丙种球蛋白的第2个疗程,可明显改善4周后运动功能结果。但目前尚缺乏充分的循证证据支持,建议根据具体临床情况个体化选择[13]。本研究中4例应用第1个疗程丙种球蛋白后出现TRF,间隔7~14 d再次给予第2个疗程免疫球蛋白治疗,肌力均有不同程度改善。本文提及的1例特殊病例1周内达运动障碍高峰,Hughes功能评分5分,予免疫球蛋白治疗第1个疗程,呼吸麻痹、意识障碍均消失,但出现显著的自主神经紊乱症状,考虑免疫损害持续存在,损害自主神经,早期的免疫治疗对现在自主功能障碍无效。因此,间隔1周后再次给予第2个疗程免疫球蛋白治疗,预后良好,随访6个月,未见复发。
研究显示,单用糖皮质激素治疗 GBS 无明确疗效,还可影响血糖、血压变化,临床上不推荐使用[1,10]。本组发现3例伴明显神经根痛,核磁共振检查提示2例有脊髓水肿/腰椎段神经根强化,给予丙种球蛋白治疗后肌力明显改善,疼痛感明显,考虑激素强大的抗炎作用,可改善炎症刺激及脊髓肿胀,均加用糖皮质激素(短期),效果显著,因无大样本研究支持,仍需进一步研究及改善。
GBS为单相病程,有自愈倾向,儿童神经再生能力更强,短期恢复行走能力优于成人[14],本12例患儿仅1例短期预后出现共济失调,余均预后良好,肌力均恢复正常。研究中发现Hughes功能评分大于3分的病例3例,均有肌力下降(Ⅱ~Ⅲ级)、脊髓水肿或神经根强化,其中1例肌肉电传导减慢(左右尺神经CMAP波幅稍低)失电位型,遗留共济失调、头晕不适,因此认为神经失电位型、Hughes功能评分大于 3分是GBS短期预后的影响因素,与孙瑞迪等[14]国内研究相一致。而脊髓病变(水肿或神经根强化)也需列入其影响因素范围,这还需进一步大样本、多中心研究论证。
总之,儿童GBS变异型发病率低,临床症状不典型,误诊率高,特别是以非肌力下降为首发症状的患儿,临床医师应重视神经专科查体及详细的病史采集,拓宽诊断思路,避免误诊。一经确诊,早期应给予免疫球蛋白治疗,如病程中出现TRF,根据个体情况,可给予第2个疗程免疫治疗。
