铜代谢相关蛋白及关联疾病研究进展
2022-05-07王天城综述杨旭龙汤依萍彭青和周安吴鹏审校
王天城(综述), 杨旭龙, 汤依萍, 彭青和, 周安, 吴鹏(审校)
铜是人类肝脏中含量较高的必需过渡金属,仅次于铁和锌,是体内多数氧化酶的辅基。铜离子作为辅助因子传递给铜蓝蛋白(ceruloplasmin,CP)、超氧化物歧化酶1(superoxide dismutase 1,SOD1)、金属硫蛋白(metallothionein,MT)、细胞色素C氧化酶(cytochrome C oxidase,COX)、凝血因子Ⅴ和Ⅰ等,参与细胞能量代谢、抗氧化、神经递质形成等重要生命活动[1]。体内铜的含量需要维持相对稳定,铜代谢发生异常会引发一系列的机体功能障碍,引起严重的临床症状。
铜进入机体后,由小肠吸收,转运至肝脏内代谢,然后通过血液运输至各组织器官。以细胞利用铜离子为例,还原成一价的铜离子由铜转运蛋白(copper transporter 1,Ctr1)转运至细胞内,二价金属转运蛋白(divalent metal transporter 1,DMT1)也可吸收少数的Cu2+。胞内铜通过超氧化物歧化酶铜伴侣蛋白(copper chaperone of superoxide dismutase,CCS)插入SOD1、通过COX17传递到线粒体中的COX合成物1/2(synthesis of cytochrome C oxidase 1/2,SCO1/2)后整合至COX中、通过抗氧化蛋白1(anti-oxidant 1,ATOX1)转运到反面高尔基体管网状结构(trans-Golgi network,TGN)上的铜转运ATP酶α肽(ATPase Cu2+transporting alpha polypeptide,ATP7A)和铜转运ATP酶β肽(ATPase Cu2+transporting beta polypeptide,ATP7B)。铜代谢结构域包含体1(copper metabolism mouse U2af1-rs1 region 1,COMMD1)参与ATP7B的细胞内运输,并且是该转运体从质膜向TGN逆行运动所必需的。适配器蛋白复合体1(adaptor protein complex 1,AP-1)调节ATP7A和ATP7B在细胞中的定位(图1)。近年来,随着分子生物学和遗传学的发展,铜代谢的基础与临床研究取得了一定进展。本研究将从铜代谢相关基因、蛋白结构、生理功能及关联疾病作一综述。
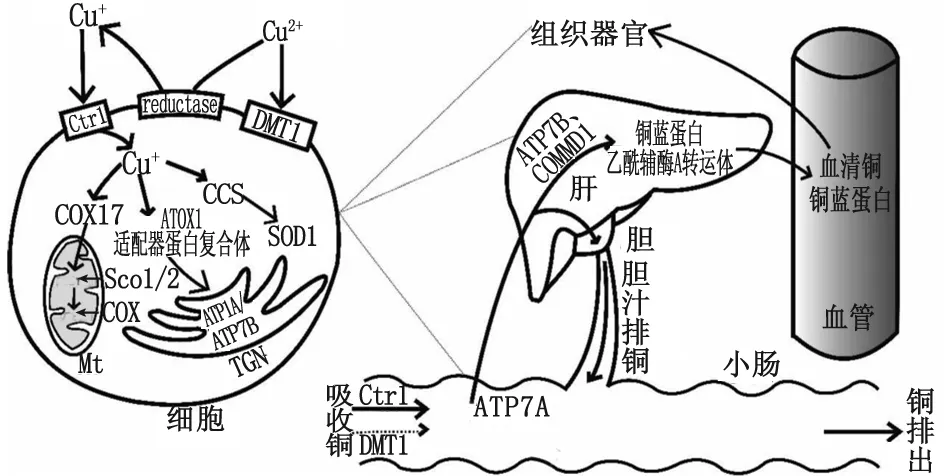
图1 铜代谢相关蛋白模式图Fig.1 Diagram of copper metabolism related proteins
1 肠道吸收铜代谢相关蛋白
1.1 Ctr1 Ctr1是一种由SLC31A1基因编码的对Cu2+具有高亲和力的转运蛋白,主要定位在细胞膜上[2]。人类铜转运蛋白1(human copper transporter 1,hCtr1)含有190个氨基酸残基,其分子结构主要由4个部分构成:(1)1个暴露在膜外的糖基化N-末端结构;(2)3个单环跨膜区域结构;(3)1个在第一和第二跨膜结构间起连接作用长度可变的细胞内环形结构;(4)1个膜内的C-末端结构。Ctr1蛋白暴露在膜外的糖基化N-末端,含有较丰富的蛋氨酸和组氨酸,具有交缠结构,有利于Cu2+的结合。
人体内铜的吸收主要依赖位于肠道细胞顶膜和大多数组织中的Ctr1,由于Ctr1只能对Cu+进行转运,所以Cu2+会先还原成Cu+,这一过程可能是金属还原酶、维生素C及DCYTB介导的。Cu+在富含蛋氨酸和组氨酸的N-末端停靠点通过Ctr1寡聚体对称的通道状结构进入细胞内[3]。Ctr1的N-端富含组氨酸的结构域,对Cu2+有着很高的亲合度并且参与Ctr1对铜的摄取。Cu+借助Ctr1蛋白从膜外主动转运至膜内后,作为辅助因子在细胞内相关蛋白间迁移。自然选择进化出了一套十分严密的调控网络,通过一系列蛋白确保Cu+安全到达正确的靶点而不产生危害[4]。目前研究发现,hCtr1蛋白捕获的Cu+主要传递至3个靶向蛋白:线粒体中的COX、细胞质中的SOD1和TGN。在动物模型研究中发现,Ctr1蛋白发生异常会导致胚胎死亡,致病基因为SLC31A1,但人类中未见报道[5]。
1.2 ATP7AATP7A基因定位于Xq21.1,含有23个外显子,是编码1 500个氨基酸的铜转运p型ATP酶。ATP7A的蛋白结构包括3个部分:N-端6个金属结构域(metal-binding domain 1~6,MBD1~MBD6)、8个跨膜区域(transmembrane domain 1~8,TMD1~TMD8)以及C-端区域[6]。TMD1~TMD8组成ATP7A蛋白的铜跨膜运输通道,TMD4和TMD5之间为活化区域(phosphatase domain,A-domain),TMD6与TMD7之间存在一个磷酸化区域(phosphorylation domain,P-domain)和核苷酸结合区域(ATP-binding domain,N-domain)。ATOX1传递的Cu+可与MBD1~MBD6结合,MBD通过其激活启动泵送[7]。MBD1~MBD4主要与A-domain相互作用,MBD5~MBD6则调节酶与铜的亲和力,每个MBD都有1个由柔性环连接的紧凑折叠使其行动[8]。ATP7A羧基末端存在保守的2个亮氨酸残基,ATP7A在细胞中的内吞和外排作用与之相关。
ATP7A定位于TGN上,主要功能为调控Cu2+运输、排泄以及催化ATP分解。除肝脏外的组织中,铜通过ATP7A离开细胞质并穿过基底外侧膜[7]。正常生理情况下,铜会被泵入分泌途径,ATOX1将铜运输至高尔基体,并通过ATP7A转运至TGN,促进铜酶的合成[9]。当胞内铜水平偏高时,ATP7A会重新定位,从而泵出多余的铜[8],高尔基体上ATP7A迁徙到细胞膜上,Cu2+与其N-端的金属结合位点结合后,ATP与ATP激酶完成结合并生成ADP。此时Cu2+转移至酶核心,最后通过去磷酸化将多余的铜排出细胞,当胞内铜稳态恢复后ATP7A重返至高尔基体上[9]。
ATP7A基因缺陷导致X连锁疾病:枕角综合征(OMIM 304150)、脊髓性肌萎缩远端X连锁3(SMAS3,OMIM 300489)和门克斯病(OMIM 309400)。ATP7A基因近半数突变位于外显子4、9、10和15。BAKKAR等[10]研究发现,ATP7A基因中携带p.Met1311Val(M1311V)替代变种,该突变对其作为Ctr1的作用产生负面影响,并损害运动神经元功能和形态。门克斯病是一种由ATP7A基因突变引起的X-连锁隐性遗传病,基因突变导致ATP7A功能障碍,进而引起体内铜分布异常,神经组织、肝脏和血液中的铜含量严重缺乏。门克斯病患者的临床特征可以解释为缺乏铜依赖酶[8]。该病发病率为0.000 27%~0.000 33%[11],到目前为止,已发现超过270种ATP7A基因突变可以导致该疾病的发生。FUJISAWA等[12]通过聚合酶链式反应扩增和外显子的直接测序明确诊断门克斯病,该研究调查了日本1975—2013年间的66例患者,共检测到55种不同的突变,其中20种是新突变,突变位于6个铜结合位点、第1~3和第6跨膜区以及ATP结合位点周围。毛雨鸽等[13]研究发现,铜补充剂作为门克斯病的一线治疗药物,可直接提高脑内和血清中的铜含量。侧脑室注射高剂量携带有ATP7A基因的重组腺相关病毒9型(rAAV9-sATP7A)联合肌内注射铜-组氨酸有望成为新的治疗方式,但其安全性和有效性仍有待临床试验证实[13]。
1.3 DMT1DMT1基因定位于12号染色体,含有43 999对碱基,包含17个外显子,cDNA全长4 142 bp,最初被称为自然抗性相关巨噬细胞蛋白2(natural resistance associated macrophage protein 2,Nramp2),后因研究证明其可转运Fe2+和Cu2+等离子,故又被命名为二价金属离子转运体[14]。DMT1蛋白结构包括12个跨膜结构域、膜内的氨基和羧基末端,第4跨膜区是其重要功能区域,若发生突变会严重影响DMT1的正常功能,其mRNA由不同的剪切方式产生+IRE和-IRE两种类型。
DTM1作为维持胞内二价金属离子稳态的重要转运蛋白,其基因广泛表达于小肠、肝脏、肾脏和肺等组织器官[15],主要分布在细胞内循环的内吞小泡和细胞膜上,可以转运铁、铜、锰等多种二价金属离子,发生异常所关联的疾病是小细胞低色素性贫血并肝铁过载。
2 肝脏铜代谢相关蛋白
2.1 ATP7BATP7B基因定位于染色体13q14.3,全长约85 kb,包括21个外显子和20个内含子,由1 465个氨基酸组成[16-17]。主要在肝脏和肾脏中表达,肺、胎盘和大脑中也有少量表达。ATP7B是一种跨膜蛋白,主要由3个部分构成:(1)6个N-端铜离子结合区,每个大约包含30个氨基酸,每个ATP7B分子可与6个Cu2+结合;(2)8个不连续的横跨膜离子通道,是金属离子转运区域的标记;(3)P型 ATP功能区,P-domain是P型ATP酶的标记、A-domain使ATP酶结构还原、N-domain是ATP的结合点。其中外显子2可与细胞质中游离的Cu2+结合,即位于N-端的MBDs。
ATP7B主要的生物学功能是在肝细胞中将胞质内ATOX1蛋白携带的Cu2+转运至TGN,并将铜负载到新合成的CP上,参与CP的合成。在高铜浓度下,ATP7B转运至溶酶体,将过量的铜运输至囊泡,通过胞吐作用随胆汁经胆小管排泄[15]。在此过程中,ATP水解,为ATP7B磷酸化提供能量,随后再进行脱磷酸化,为铜跨膜供能[17]。就ATP7B而言,不同突变间的致病性存在较大差异。即使是位于同一结构域的临近突变,也可能存在截然不同的功能特性。ATP7B主要基因突变、功能改变及临床表型见表1。
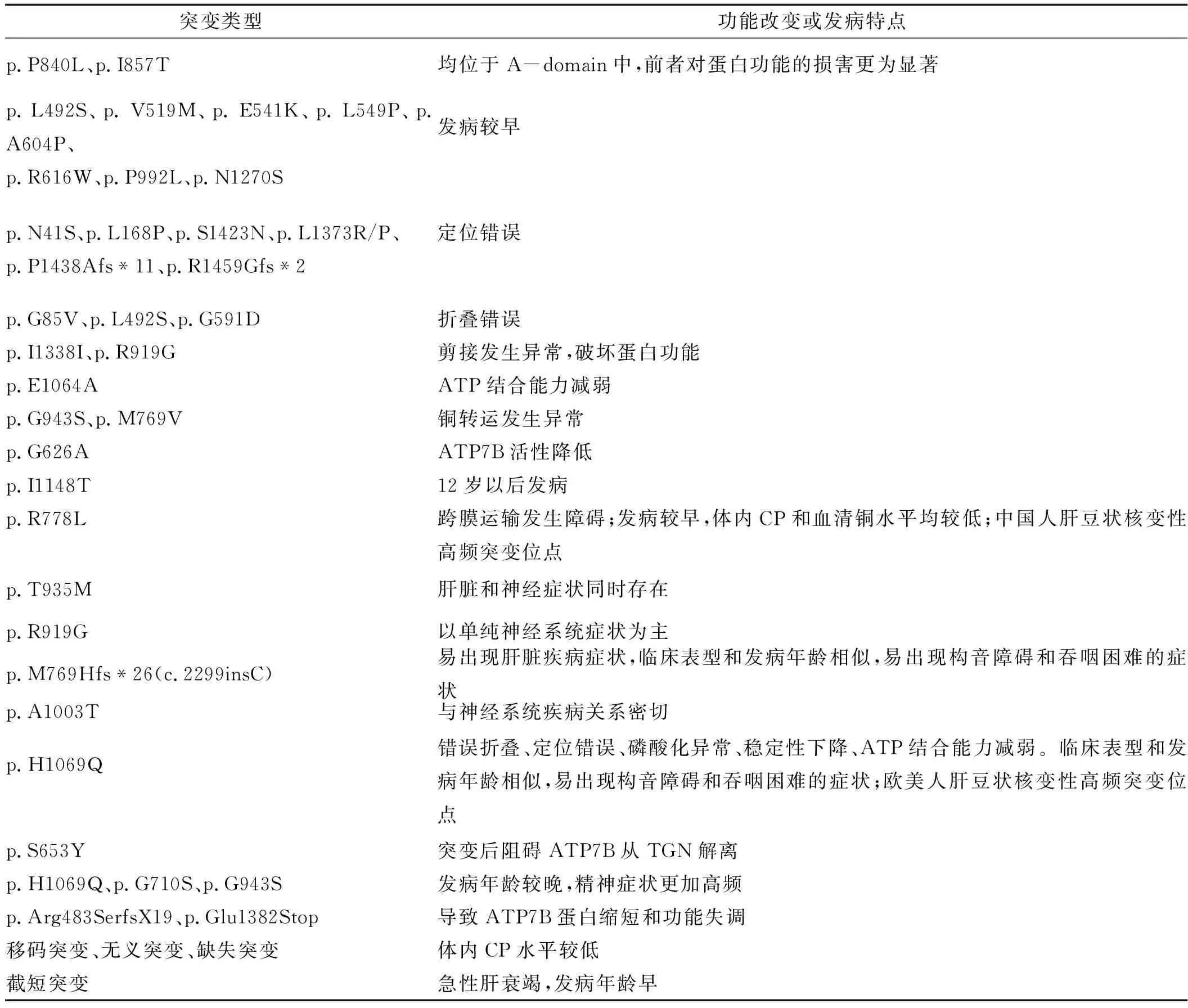
表1 ATP7B突变、功能改变及临床表型Tab.1 ATP7B mutation,functional changes and clinical phenotype
ATP7B发生突变后可引起肝豆状核变性(hepatolenticular degeneration,HLD),又称Wilson病(wilson’s disease,WD),是一种常染色体隐性遗传病。ATP7B突变后无法与Cu2+结合,导致铜在肝脏、大脑等组织器官中形成系统性沉积,继而引起各种临床表现。主要表现为进行性加重的肝硬化、锥体外系症状、精神症状、肾损害及角膜色素环(kayser-fleischer ring,K-F环)等,以进行性肝病和神经精神障碍最常见。WD患者因ATP7B功能异常,导致高尔基体中CP无法承载铜,因此无法适当地将铜排泄到胆汁中[18]。《中国肝豆状核变性诊治指南2021》[19]简化了HLD的诊断标准,指出临床上对于高度怀疑患者筛查ATP7B基因是否发生p.R778L、p.P992L和p.T935M 3种突变类型的重要性,强调ATP7B基因筛查对于“可能肝豆状核变性”患者的重要性。目前,WD的治疗核心主要是抑制铜的吸收和促进铜的排泄。指南强调出现症状前的个体以及经过治疗症状稳定的患者的维持治疗,可单用锌剂或者联合应用小剂量络合剂。指南同时指出,肝移植术后患者应坚持低铜饮食并口服锌剂,并对D-青霉胺、二巯丙磺酸钠、二巯丁二酸胶囊及锌制剂的用量作出了新的规范要求。ZHOU等[20]通过随机对照试验比较WD患者在用金属质子治疗期间大脑磁敏感加权成像变化的方法,认为与D-青霉胺相比,使用二巯丙磺酸钠和二巯丁二酸的治疗可更快改善WD患者的神经症状,减少恶化概率,为临床用药提供指导。CAI等[21]通过对WD患者的粪便样本进行16S rRNA测序,并与健康人群进行比较,发现WD患者肠道微生物群的多样性和组成明显低于健康个体以及前组宿主免疫和代谢相关系统通路所涉及的菌群数量较低的现象,这为未来从肠道微生物菌群着手治疗WD提供了新的思路。汤珊等[22]研究发现,CRISPR/Cas9介导的ATP7B点突变校正是可行的,有可能应用于临床治疗WD。VTX-801是一种携带小型功能性ATP7B铜转运体基因的腺相关病毒药物,其治疗机制在于通过外源性导入铜转运体基因来恢复肝细胞的铜代谢水平。2021年8月,Vivet Therapeutics公司和辉瑞公司共同宣布VTX-801获得美国食品药品监督管理局快速通道资格。目前VTX-801基因疗法已在临床前模型中得到了验证,并已获得美国食品药品监督管理局和欧盟委员会的孤儿药资格认定[22],这是WD基因治疗药物的一项重大进展。
2.2 COMMD1 人类COMMD1基因定位于染色体2p13-16,全长约235 kb,包括3个外显子和2个内含子。COMMD1蛋白质包含190个氨基酸,具有代表性的COMMD结构域位于残基C-端,N-端含有多个α-螺旋结构。该蛋白在多种组织中均有表达,肝脏中与ATP7B的N-端直接相连。
COMMD1是COMMD蛋白家族中最典型的成员,是一种多功能性因子,参与许多生理过程的调节,包括铜稳态、离子转运、氧化应激、蛋白质运输、NF-κB介导的转录、缺氧诱导的转录、DNA损伤反应和肿瘤发生等。COMMD1调节ATP7A和ATP7B的稳定性,诱导突变或错误折叠的转运体泛素化和蛋白酶体降解,突变导致门克斯病和WD,其第164位天冬氨酸的同义突变p.D164D(c.492T>C)是ATP7BH1069Q纯合突变患者神经和肝脏症状早发的重要因素。COMMD1基因变异已被证明会导致伯灵顿犬发生铜中毒,其临床表现与人类WD相似[5]。
3 血液转运铜相关蛋白
3.1 CP CP又称铜氧化酶,是一种含铜的α2糖蛋白。人体CP基因定位于8号染色体3q24-q25.1,基因长度为45~65 kb,包含19个外显子,由1 046个氨基酸和附着的4个寡糖-氨基葡萄糖组成,其相对分子质量约为132 kD。CP以单一多肽链的形式存在于人体中,完整的单一多肽链自动裂解后形成3组由结构域A1、A2、B组成的异体同形单元。每单元约有350个氨基酸残基,3组单元间氨基酸序列高度同源。血浆中CP可结合6个铜原子,一、六区的交界面上的3个铜原子形成三核铜簇,对CP的催化活性和结构稳定有着重要作用,其余3个铜原子以单核形式存在于二、四、六区。CP属于多铜氧化酶家族,具有氧化酶功能,可将Cu+氧化为Cu2+,使其在体内进行转运与代谢。XU等[23]研究发现,当CP水平<20 mg/dL时,WD的敏感性为99%、特异性为80.9%,CP水平<15 mg/dL时,WD的敏感性为95%、特异性为95%。此外CP与WD、遗传性铜蓝蛋白缺乏症(hereditary aceruloplasminemia,HA)以及阿尔茨海默病(alzheimer disease,AD)等密切相关。
HA是一种罕见的常染色体隐性遗传病,CP是其致病基因,主要临床表现为血色病、胰岛素依赖型糖尿病、视网膜变性、锥体外系症状、皮质下痴呆等神经系统退行性变性症状,多数有轻度贫血[24]。Cu2+结合结构域发生突变,会导致铜结合能力下降;糖基化修饰区的氨基酸发生突变,会导致CP滞留内质网;框移突变及无义突变,会导致CP表达量下降;氧化酶活性区域的氨基酸发生点突变,会导致CP亚铁氧化酶活性下降[5,24]。CP发生错义突变,会致使其与铜的结合能力缺失,如Q692K、D58H、G969S和G631R等突变。AD是一种常见的中枢神经系统退行性疾病。邓青芳等[25]的研究发现,AD与CP异常相关,血液中游离铜水平与AD患者的认知能力成负相关,并可预测认知能力丧失的速度。AD患者脑脊液中有活性的CP减少,非活性的铜增加,这一病理变化可能对AD的发生发展起一定作用。
3.2 乙酰辅酶A转运体 乙酰辅酶A转运体主要功能是CP乙酰化修饰,其致病基因为SLC33A1。发生异常时会出现低铜、CP缺乏及铜缺乏等症状,与之相关联的疾病是Huppke-Brendl综合征[5]。该综合征是一种致死性的常染色体隐性遗传病,伴有严重的发育迟缓、听力丧失以及先天白内障,患者的脑MRI表现为明显的小脑发育不全和髓鞘形成不良,与门克斯病相似。
4 组织细胞利用铜相关蛋白
4.1 AP-1 AP-1定位于染色体7q22.1上的AP1S1基因,相对分子质量为100 kD,编码受体分子上起连接信号蛋白作用的小S亚单位(σ1A亚基)[26]。AP-1定位于TGN和内体,与网状蛋白结合参与ATP7A、ATP7B从TGN和内体向质膜运输及网状蛋白外壳的组装[27]。
MEDNIK综合征是由AP1S1基因的7q22.1位点发生隐性突变引起的一种罕见的常染色体隐性神经皮肤遗传疾病,此时AP-1介导的细胞内转铜蛋白异常转运,ATP7A和ATP7B有完整的酶活性,但功能发生障碍,导致铜代谢功能障碍、铜运输不足以及铜依赖酶减少[26]。AP1S1调节ATP7A在细胞内正确定位的发现,首次证明MEDNIK综合征是一种铜代谢紊乱相关疾病。结合门克斯病和WD在铜代谢异常方面的表型特征,MEDNIK综合征的临床表现为角化病、鱼鳞病、肠病、听力丧失、智力低下和周围神经病,新生儿从出生到1岁间即可表现出相关临床症状[26]。MEDNIK综合征可以将表现出复杂的神经皮肤表型、肠道疾病的临床评估和家族史调查作为诊断辅助,建议在鱼鳞病和智力残疾儿童的鉴别诊断中考虑进行AP1S1基因分析。
4.2 ATOX1 人体ATOX1基因(human ATX1 homologue,HAH1)定位于染色体5q32-5q33,全长502 bp。编码的ATOX1是含68个氨基酸的多肽,经加工形成具有稳定结构的单链蛋白质,含2个α螺旋、2个β折叠和1个N-端MXCXXC的结构域,其N-末端的两个半胱氨酸残基负责结合Cu+。
HAH1与ATOX1有着相似的抗氧化活性,均可将Cu+转运给具有抗氧化作用的相关蛋白,但自身却无抗氧化功能。位于hCtr1 C-端8肽的Cu+被转移给ATOX1[28],ATOX1与之结合后,通过利用ATP水解释放出的能量将铜转运至TGN上ATP7A和ATP7B的-NH2末端,参与合成各种铜依赖酶。
ATOX1通过变构调节MBDs启动ATP7A/B泵的活性[8]。在体外实验中,纯化的ATOX1铜结合能力随着时间的推移而减弱[29]。GE等[30]发现,ATOX1与CCS的结构域Ⅰ在体外的磁共振波谱相互作用下促进铜的交换。ATOX1代谢紊乱时,会引起系统性铜缺乏,严重时会导致胚胎死亡或哺乳期夭折[1]。
4.3 CCSCCS基因定位于染色体11q13.2,由274个氨基酸残基组成[30]。CCS蛋白含有249个氨基酸,相对分子质量约29 kD。CCS单体有3个不同作用的结构域:(1)结构域Ⅰ位于N-端,由88个氨基酸组成,N-端含一个MXCXXC金属结构域(铜结合位点),类似于ATOX1;(2)结构域Ⅱ位于中间,与SOD1高度同源但无SOD活性;(3)结构域Ⅲ位于C-端,是由40个氨基酸残基组成的一段无规则卷曲的多肽链,C-末端含有分子内S-S桥形成所需的铜催化CXC位点[8,31]。
CCS是存在于细胞质中SOD1的铜伴侣分子,能将Cu+专一性地插入SOD1的铜结合位点,促进形成二硫键介导激活SOD1,Cu2+位于SOD1的活性中心,通过得失电子实现SOD1的歧化作用[32]。SOD1活性在蛋白水平上受铜调控,而SOD3活性在基因水平上受铜调控[8]。细胞正常增殖发育必须依赖SOD1的抗氧化作用,因此通过抑制CCS的表达实现抑制肿瘤细胞的增殖。CCS的缺乏会导致SOD1发生缺陷、活性下降,甚至是CCS缺乏症[5]。HATORI等[29]指出,铜缺乏时CCS上调与ATOX1交换、分配铜,供SOD1成熟。
4.4 COX17COX17功能基因定位于3号染色体,假基因定位于17号染色体。COX17编码的酸性蛋白定位在胞质与线粒体内膜之间,含有69个氨基酸残基,相对分子质量为8.2 kD。COX17的C-端含有线粒体结合位点,可将由Ctr1转运至胞内的Cu+转运至线粒体,通过线粒体膜上的SCO1/2将Cu+组装到位于呼吸链末端传递电子的COX中。COX17以未折叠的形式进入线粒体膜间隙,从线粒体腔接受铜,并向COX11输送铜。Cu2+是COX的活性中心,通过参与呼吸链电子的传递产生能量[26]。COX17代谢紊乱会使COX活性下降[5],缺失可导致呼吸链缺陷,严重可导致胚胎死亡[1]。
5 总结与展望
铜作为机体必须的微量元素,在人体正常生命活动中发挥着重要作用,但同时具有一定毒性,超载或缺乏都会引起相关疾病。研究生理状态下铜代谢的相关基因、蛋白表达、生理功能,有助于了解临床上与之关联的疾病,为临床治疗提供方向。目前与铜代谢相关蛋白相关的问题主要集中于以下两点:一是可能存在尚未被发现的铜代谢相关蛋白;二是关于已发现的相关蛋白的研究仍有新突破可能,如CP在AD发生发展过程中的作用机制,胚胎期Ctr1发生异常所导致的疾病等。铜代谢的研究与发展前景十分广阔,一方面利用一些新技术、新手段,如Duolink®PLA®技术、lcp结晶和微晶电子衍射(Microed)可能会发现新的基因突变位点,为临床治疗提供新的靶点。另一方面针对已发现的铜代谢相关蛋白的研究,主要分两个层次,一是因基因突变引起的铜代谢疾病可采取基因治疗,但受限于伦理;二是非基因突变引起的铜代谢疾病存在突破可能。今后可进一步研究两者关系,将铜代谢的基础研究与关联疾病的临床实践相结合,探讨致病机制,采取针对性治疗手段,以提高临床疗效。
