拟南芥HKT1基因pCAMBIA载体构建及转基因材料筛选
2022-05-06徐洁娜夏明慧樊婷婷
徐洁娜, 夏明慧, 樊婷婷
(合肥工业大学 食品与生物工程学院,安徽 合肥 230601)
0 引 言
随着地球水资源的不断减少,土壤盐渍化程度越来越严重,高盐环境对植物的生长有着极其严重的影响,从而影响全球农作物产量[1-2],因此找到响应盐胁迫关键的转录因子,以及参与到信号通路的下游基因十分重要[3-4]。利用转基因技术优化植物品种以达到抗盐目的是目前解决以上问题较为经济快捷的方式[5]。
细胞质中过量游离性的Na+对植物细胞有害,因此植物应该严格限制Na+的吸收、长距离运输和积累[6]。已知编码高亲和力K+转运体的基因HKT1,介导Na+从木质部到木质部薄壁细胞的运输,减少了木质部Na+含量,大大提高了植物的耐盐性[7]。目前,关于HKT1耐盐的机理已明确,但是其上游调控机制尚不明确。现已知MYBX是一个响应盐胁迫的关键转录因子,其功能缺失突变体对150 mmol/L NaCl高度敏感。初步分析发现基因HKT1在突变体mybx中表达量明显降低,在过表达植株中显著上升。因此,本文推测MYBX基因正向调控HKT1基因的表达,需要探索其基因上下游的关系,构建HKT1的过表达载体,获得1300-HKT1表达菌株,成功侵染mybx突变体,得到1300-HKT1/mybx转基因材料。
1 材料与方法
1.1 实验材料
1.1.1 材料
模式植物拟南芥(Arabidopsisthaliana)、野生型Col-0和突变体mybx,购于美国拟南芥种质资源中心。
1.1.2 宿主和载体
大肠杆菌DH5α、农杆菌GV3101、pCAMBIA1300质粒。
1.1.3 分子生物学试剂
Trizol(RNA提取试剂)、PrimeSTAR酶(高保真DNA聚合酶)、反转录试剂盒RevertAid First Strand cDNA Synthesis Kit、质粒提取试剂盒Plasmid Mini Kit 50-preps、OMEGA琼脂糖胶回收试剂盒Cycle-Pure Kit(购于北京安诺伦生物科技有限公司);DNA supermix(DNA聚合酶,生工牌)、限制性核酸内切酶(NEB牌)、T4连接酶(购于TaKaRa公司);合成引物(Oligo软件设计)由生工生物工程(上海)股份有限公司;其余试剂为分析纯。
1.2 拟南芥无菌苗的培养
从4 ℃冰箱取出MS(Murashige & Skoog)培养基,恢复至室温,以100 mL的培养基为例。称取0.435 g MS培养基,蔗糖0.8 g,琼脂粉1.2 g。加入ddH2O混匀后,调pH值至5.8,高温灭菌,倒入平板等其凝固。在无菌操作台内使用1 mL 0.1%的HgCl2溶液重悬拟南芥种子,时间控制在3 min内,然后用无菌的ddH2O重悬种子3次,弃上清,将无菌的拟南芥种子转移至无菌滤纸上,待其干燥后,按照实验需求播种在培养基上。播种结束后,用封口膜将培养皿密封保存于4 ℃冰箱,春化3 d后放置于光照培养间竖直培养14 d。
1.3 植物RNA的提取及反转录
提前洗净研钵放置60 ℃烘箱烘干后,用锡箔纸包裹置于180 ℃烘箱3~6 h,再转移至冰柜预冷待用。所用试剂氯仿、异丙醇、无水乙醇也同时放置冰箱预冷。收集培养皿中的无菌苗,置于预冷的研钵中,加入液氮研磨至粉末发白,加入1 mL Trizol,继续研磨,直至混合物变成清亮的液体,转移至预先标记好的1.5 mL离心管中,冰上静置5 min,接着13 000 r/min、4 ℃离心10 min;吸取上清至新的离心管中,加入200 μL预冷的氯仿,剧烈震荡15 s,静置5 min,接着13 000 r/min、4 ℃离心5 min;吸取上清至新的离心管中,沿壁缓缓加入500 μL预冷的异丙醇,轻柔上下颠倒数次,冰上静置15 min,13 000 r/min、4 ℃离心15 min;小心弃掉上清,再一次沿壁缓缓加入1 mL 75%的乙醇,8 000 r/min、4 ℃离心5 min,弃上清,沉淀即为所提RNA。室温晾干,加入50 μL DEPC水,65 ℃水浴10 min,期间轻柔吹打液体使其充分溶解。取2 μL RNA,测得RNA质量浓度和纯度后,根据检测结果调整RNA的加样量,尽量保证其质量浓度一致。依据Thermo Scientific公司提供的RevertAid First Strand cDNA Synthesis Kit反转录试剂盒反转合成cDNA。
1.4 载体构建
在TAIR网站(https://www.arabidopsis.org/)内找到拟南芥HKT1基因序列(TAIR:AT4G10310),利用Oligo引物合成软件来设计HKT1编码区的上下游引物。根据pCAMBIA1300载体酶切位点信息以及基因本身序列信息,选择XhoⅠ和HindⅢ这2个限制性酶切二位点。将这2个酶切位点碱基序列分别加在上下游引物的5’端。上游引物为:
5’-CCGCTCGAGATGGACAGAGTGGTGGCAAAA-3’,
下游引物为:
5’-CCCAAGCTTTTAGGAAGACGAGGGGTAAAG-3’。
以拟南芥野生型(WT)的cDNA为扩增模板,通过聚合酶链式反应(polymerase chain reaction, PCR),获得HKT1基因片段,凝胶回收PCR产物,取38 μL片段或质粒,加入相应的限制性核酸内切酶1 μL(动作迅速且在冰上操作)和5 μL CutSmart,最后用ddH2O补齐至50 μL体系,吹打混匀后,放置于37 ℃金属浴中孵育,目的基因片段酶切2 h,质粒酶切4 h。凝胶回收后,酶切片段和酶切载体相连以转化DH5α 感受态细胞,并鉴定阳性克隆(命名为1300-HKT1)用于测序。测序成功后的大肠菌株进一步扩培,利用质粒抽提试剂盒获取目的质粒,载体构建成功。
1.5 农杆菌转化
提前准备预冷的电击杯,取出-80 ℃冻存的GV3101,加入1 μL重组质粒,轻混匀后转移至电击杯中,设置电转仪参数,电脉冲25 μF,电压1 800 V,启动电脉冲。经过电击后,迅速取出电击杯,向其中加入500 μL预冷的液体培养基,28 ℃、200 r/min振荡培养4 h。待LB浑浊后,在无菌操作台内将菌液转移至含有100 mg/mL卡那霉素抗性的固体培养基上,吹干,30 ℃培养箱密封培养2 d。待农杆菌菌落直径约为2 mm时,即可挑取单克隆至500 μL相应抗性液体培养基中,28 ℃、200 r/min振荡培养,24 h后PCR鉴定,通过凝胶电泳结果选取活性较高的菌落保菌,存于-80 ℃待用。
1.6 浸花法侵染拟南芥
土培繁殖mybx突变体,待其生长至6周左右,顶端分生组织发育旺盛、花苞较多,正是侵染构建转基因材料的最佳时期。提前配制MS/蔗糖侵染缓冲液。活化1300-HKT1农杆菌:从-80 ℃冰箱中取出甘油菌,吸取100 μL菌液于5 mL含有50 mg/mL庆大霉素和100 mg/mL卡那霉素的双抗性液体培养基中,28 ℃、200 r/min过夜活化;在无菌操作台内吸取2 mL过夜活化的菌液于100 mL 同样的双抗性液体培养基中,28 ℃、200 r/min培养菌液至OD600=1.0~1.5。5 000 r/min、5 min离心收集菌体,侵染缓冲液重悬沉淀菌体3次,最后稀释管底菌体至OD600=0.5~1.0,取10 mL菌液加入0.1 μL的Silwet-77,将其混匀后静置避光待用。
处理待侵染植株方法如下:剪去突变体材料已授粉的花及果荚,将剩下未授粉的花苞浸入侵染液中15 s,保证植株每个未授粉的花苞都被侵染液润湿;整盆拟南芥侵染完用保鲜膜将整棵植株包裹,保持其湿润度,黑暗处理过夜;2 d后,小心去除保鲜膜恢复光照;1周后再次侵染。2周左右果荚陆续成熟,此时可收集种子并放入37 ℃的培养箱进行干燥处理,随后转移至4 ℃冰箱春化,筛选待用。
1.7 转基因材料筛选
提前配制含有20 mg/mL潮霉素的1/2 MS固体培养基,方法与本文拟南芥无菌苗培养方法中固体培养基配制方法一致。在无菌操作台内将消过毒的种子均匀地洒在培养基上,封口密封保存于冰箱4 ℃春化3 d。由于潮霉素见光易分解,转移至光照培养间初期需要黑暗处理3 d,然后正常光照培养12 d。挑选子叶大而绿根且长的无菌苗,移至土培,保鲜膜包裹整个花盆以营造一个稳定的微环境供转基因苗生长,直至无菌苗长出新的子叶,方可揭掉保鲜膜。14 d后提取叶子的DNA进行PCR分子鉴定。
2 结果与分析
2.1 目的片段的PCR扩增
利用生长2周的无菌苗所提RNA反转出的cDNA为模板,通过PCR扩增出目的基因HKT1的CDS序列,该序列长度为1 521 bp,凝胶电泳结果如图1所示。
由图1可知,条带大小与基因片段长度相吻合。利用琼脂糖凝胶回收试剂盒回收HKT1基因片段用于后续实验。
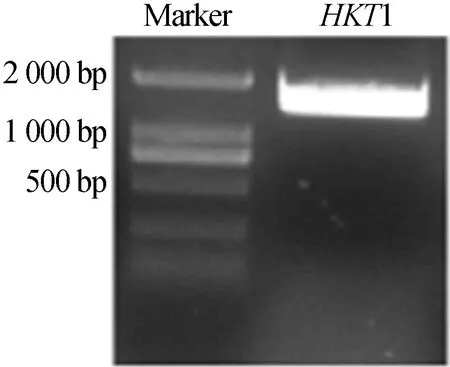
图1 拟南芥HKT1基因片段的克隆
2.2 目的基因与pCAMBIA1300质粒的双酶切
各取38 μL回收纯化的HKT1基因片段和空载质粒pCAMBIA1300分别加入限制性核酸内切酶XhoⅠ和HindⅢ进行双酶切,酶切后电泳结果如图2所示。
从图2可以看出,酶切效果良好,无拖带弥散杂带情况,目的条带清晰,且大小符合基因长度。接着使用琼脂糖凝胶回收试剂盒回收酶切产物,利用T4-DNA Ligase 将酶切后的基因片段和pCAMBIA1300质粒在16 ℃的金属浴中连接14 h,获得连接产物。

图2 基因和质粒双酶切
2.3 大肠杆菌重组质粒阳性菌落的PCR鉴定
将连接产物通过化学转化法转入DH5α感受态细胞中,涂布于卡那霉素的单抗性LB固体培养基上,37 ℃培养箱过夜培养后挑取单菌落,液体培养基培养后通过PCR鉴定出阳性菌落。大肠杆菌重组质粒阳性菌落PCR鉴定的琼脂糖凝胶电泳结果如图3所示。从图4可以看出,条带大小与目的基因大小基本一致。经过测序比对,成功得到含有目的基因的重组质粒载体。
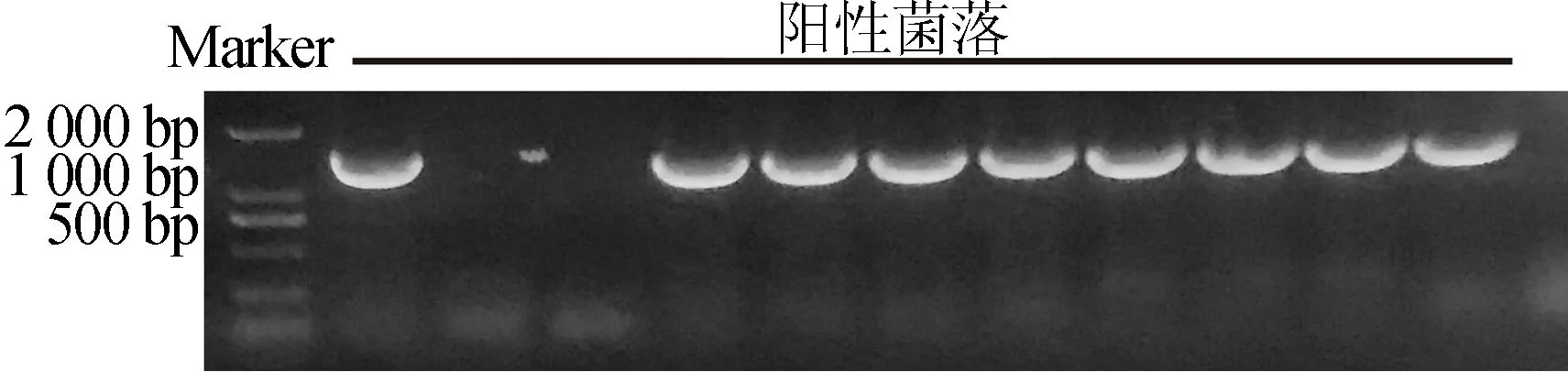
图3 大肠杆菌菌落PCR验证
2.4 农杆菌阳性菌落的PCR鉴定
用质粒小提试剂盒提取测序正确的大肠杆菌重组质粒,将正确质粒通过电转法转入GV3101农杆菌感受态细胞中,涂布于相应抗性的的LB固体培养基上,28 ℃培养2 d后,挑取单克隆于液体培养基中培养。经菌落PCR鉴定,由琼脂糖凝胶电泳结果如图4所示。从图4可以看出,条带大小与目的片段大小基本一致,说明重组质粒成功转入农杆菌中,可进行下一步突变体植株的侵染实验。
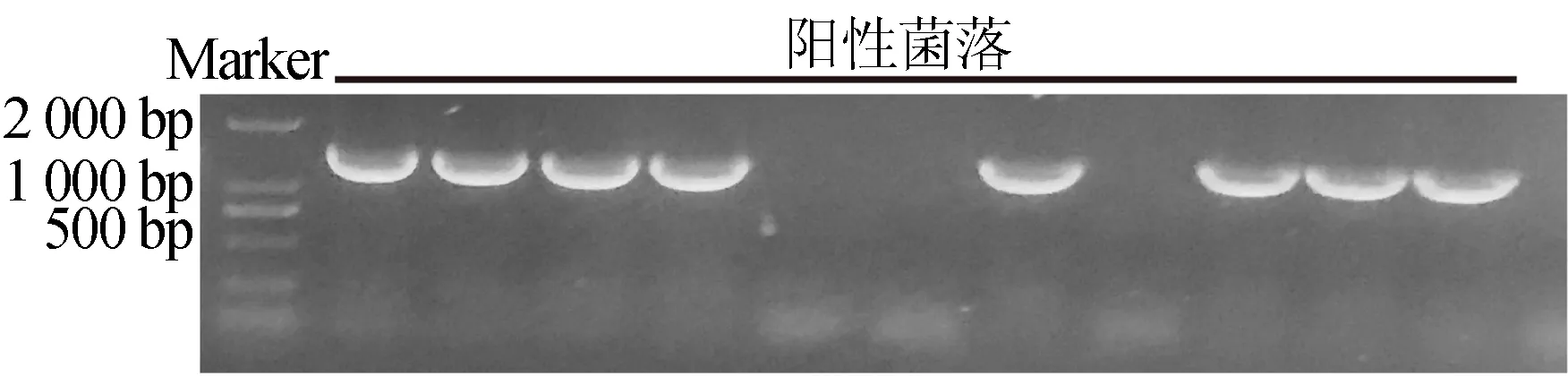
图4 农杆菌菌落PCR验证
2.5 转基因阳性植株的抗性筛选
通过浸花侵染法侵染mybx突变体收到的种子,播种在含有20 mg/mL潮霉素的LB抗性固体培养基上。2周后,阳性植株会正常生长,未侵染成功的植株因不携带潮霉素抗性基因而无法在含有潮霉素抗性的培养基上生长,表现出黄化甚至不发芽的状态,如图5所示。随后将阳性苗转移至土壤中培养。

图5 转基因阳性植株抗性筛选
2.6 转基因阳性植株的分子鉴定
为了进一步确定筛选出的拟南芥幼苗是实验所需的1300-HKT1/mybx转基因材料,并提取其莲座叶的DNA进行分子鉴定,结果如图6所示。由图6可知,凝胶电泳图中条带大小与基因片段大小一致,证明1300-HKT1/mybx转基因材料构建成功,可用于后续实验。
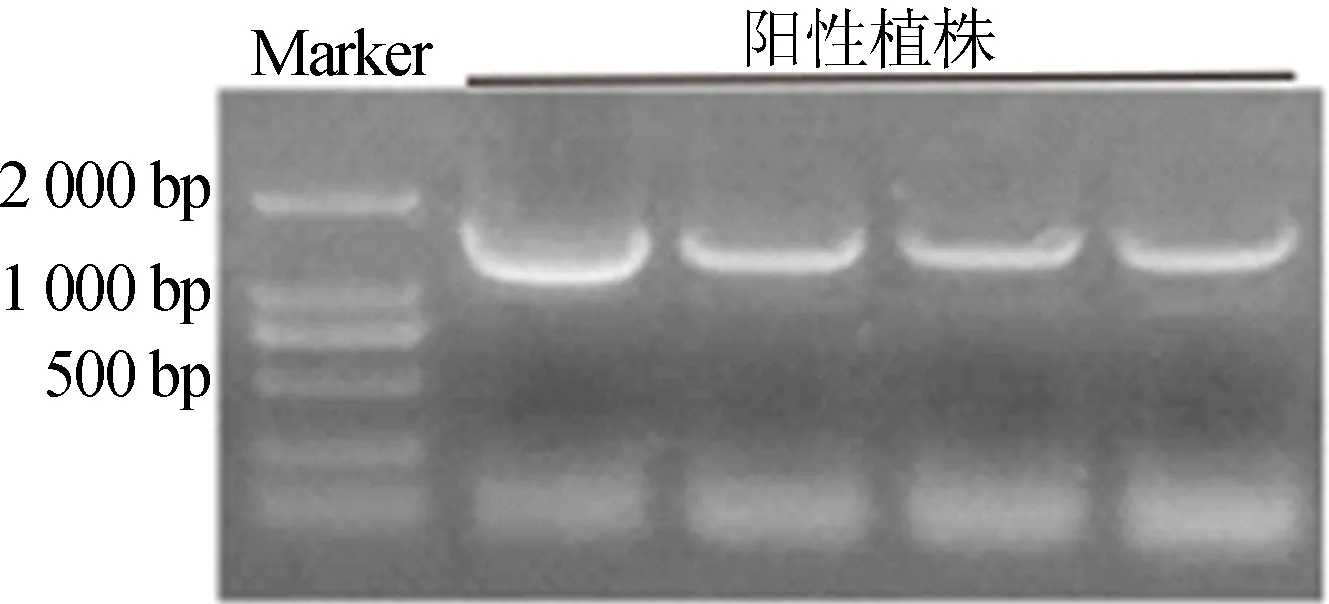
图6 转基因阳性植株分子鉴定凝胶电泳图
3 讨 论
迄今为止,全球范围内盐碱化土地约有950×104hm2,我国的盐碱化土地面积约[8]为270×104hm2。当土壤中的可溶性盐超标,就会影响土壤上植物的生长。盐胁迫包括渗透胁迫和离子胁迫以及由此引起的次级胁迫(如氧化胁迫)等一系列复杂的相应机制可能会造成植物生长滞缓,甚至死亡[9]。而我国经济型农作物多为盐胁迫敏感性植物,因此,合理开发和利用盐渍化土壤资源的根本途径,便是利用基因工程技术培育耐盐性作物品种[8]。而植物的盐胁迫响应属于多基因控制的复杂性状,因此构建相应的盐胁迫转录调控网络有利于找出各信号途径内部的基因表达调控关系,找到盐胁迫环境下的关键信号传导途径有助于揭示植物的耐盐胁迫机理[10]。因为转录因子MYBX响应盐胁迫信号,并且正向调控HKT1基因的表达,从而使得植物对高盐环境耐受,所以MYBX-HKT1信号通路机制,为植物抗盐方向提供了新思路。
